Chapter 10 Minigraph-Cactus
10.1 Cactus
https://github.com/ComparativeGenomicsToolkit/cactus
- Reference-free whole genome graph
- Constructs graph based on MSA or graph
10.2 Cactus Graphs
Cactus Graphs “naturally decompose the common substructures in a set of related genomes into a hierarchy of chains that can be visualized as two-dimensional multiple alignments and nets that can be visualized in circular genome plots”
https://www.liebertpub.com/ doi/abs/10.1089/cmb.2010.02 52
10.3 Cactus Algorithm
- Multiple sequence aligner
- Originally developed for multi-species alignments
- Fast because it uses a guide tree (Newick format)
- Now supports minigraph GFA in place of guide tree for pangenome alignments
10.4 Minigraph-Cactus
https://github.com/ComparativeGenomicsToolkit/cactus/blob/master/doc/pangenome.md https://doi.org/10.1038/s41587-023-01793-w

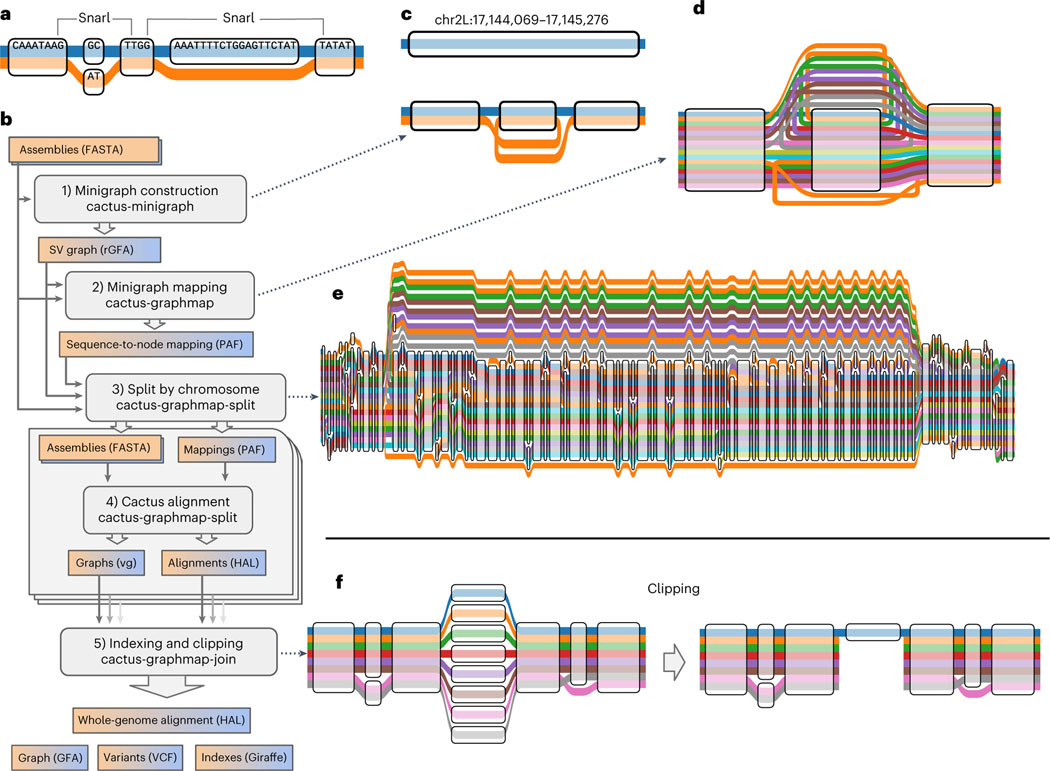
10.5 Building the Graph
10.5.1 Preparing the Input (exercise)
- Cactus seqFile tells Cactus where to load sequences from
- Maps sequence names to file paths
- We’re using:
“<strain name>\t<path to sequence>”
E.G.
seqFile: S288C ./yprp/assemblies/S288C.genome.fa
- Call it yprp.seqfile.txt
10.5.2 Singularity
https://docs.sylabs.io/guides/latest/user-guide/
- Secure containers for reproducible scientific computing
- Any user can run a singularity container
- Containers are portable, self-contained environments with everything a program needs to run
Singularity containers are saved in .sif files.
.sif files for this workshop are stored in /home/data/pangenomics-2503/sif/.
10.5.3 Cactus
Build a graph containing all of the YPRP genomes (10min):
singularity run --no-privs /home/data/pangenomics-2503/sif/cactus.sif cactus-pangenome \
./js \
./yprp.seqfile.txt \
--outDir yprp-cactus \
--outName yprp-cactus \
--reference S288C \
--giraffe \
--gfa \
--gbz \
--mgCores 10 \
--mapCores 10 \
--consCores 10 \
--indexCores 10- singularity run –no-privs /home/data/pangenomics-2503/sif/cactus.sif
- run the Cactus container with singularity
- cactus-pangenome
- the command to run inside the container
- ./js
- a job store directory where intermediate files should be stored (shouldn’t already exist)
- ./yprp.seqfile.txt
- the text file mapping sequence names to file paths
- –outDir yprp-cactus
- a directory where output files should be stored (shouldn’t already exist)
- –outName yprp-cactus
- the prefix that every output file should be given
- –reference S288C
- which input sequence should be treated as the reference
- –giraffe
- files for the giraffe read mapper should be generated
- –gfa
- a file containing the graph in GFA format should be generated
- –gbz
- a file containing the graph in GBZ format should be generated
- –mgCores 10
- minigraph should use 10 CPU cores
- –mapCores 10
- the mapper should use 10 CPU cores
- –consCores 10
- the Cactus consolidate algorithm should use 10 CPU cores
- –indexCores 10
- the indexer should use 10 cores
10.6 Read Mapping and Variant Calling
10.6.1 Reading Mapping
Map reads (10min):
vg giraffe \
-p \
-Z ./yprp-cactus/yprp-cactus.gbz \
-m ./yprp-cactus/yprp-cactus.d2.min \
-d ./yprp-cactus/yprp-cactus.d2.dist \
-f ./yprp/reads/SK1.illumina.fastq.gz \
-t 10 > yprp-cactus.SK1.illumina.gam- -p
- show progress
- -Z ./yprp-cactus/yprp-cactus.gbz
- use the Graph Burrows-Wheeler Transform generated by Cactus
- -m ./yprp-cactus/yprp-cactus.d2.min
- the minimizer index generated by Cactus
- -d ./yprp-cactus/yprp-cactus.d2.dist
- the distance index generated by Cactus
- -f ./yprp/reads/SK1.illumina.fastq.gz
- the reads to map
- -t 10
- number of threads to use
- > yprp-cactus.SK1.illumina.gam
- write the standard out (the mapped reads) into yprp-cactus.SK1.illumina.gam
Computing mapping stats (2min):
vg stats -a yprp-cactus.SK1.illumina.gam- -a
- input is an alignment (GAM) file
10.6.2 Calling Graph-Supported Variants
Compute read support for variation already in the graph (6min):
vg pack \
-x yprp-cactus/yprp-cactus.d2.gbz \
-g yprp-cactus.SK1.illumina.gam \
-Q 5 \
-s 5 \
-o yprp-cactus.SK1.illumina.pack \
-t 10- -x yprp-cactus/yprp-cactus.d2.gbz
- use the Graph Burrows-Wheeler Transform generated by Cactus
- -Q 5
- ignore mapping and base quality < 5
- -s 5
- ignore the first and last 5pb of each read
- -o yprp-cactus.SK1.illumina.pack
- the output pack file
- -t 10
- use 10 threads
Compute snarls in graph structure (<1min):
vg snarls yprp-cactus/yprp-cactus.d2.gbz -t 10 > yprp-cactus.snarls- yprp-cactus/yprp-cactus.d2.gbz
- use the Graph Burrows-Wheeler Transform generated by Cactus
- -t 10
- use 10 threads
- > yprp-cactus.snarls
- write the standard out (the snarls) into yprp-cactus.snarls
Generate a VCF from the support (<1min):
vg call \
yprp-cactus/yprp-cactus.d2.gbz \
-k yprp-cactus.SK1.illumina.pack \
-r yprp-cactus.snarls \
-t 10 > yprp-cactus.SK1.illumina.graph_calls.vcf- yprp-cactus/yprp-cactus.d2.gbz
- use the Graph Burrows-Wheeler Transform generated by Cactus
- -k yprp-cactus.SK1.illumina.pack
- use the pack file we just generated
- -r yprp-cactus.snarls
- use the snarls file we just generated
- -t 10
- use 10 threads
- > yprp-cactus.SK1.illumina.graph_calls.vcf
- write the standard out (the called variants) into yprp-cactus.SK1.illumina.graph_calls.vcf
10.6.3 Calling Novel Variants
Augmenting a single graph can take much more time than making a separate graph for each chromosome and augmenting them individually. Make a separate graph for each chromosome (1min):
vg chunk -x yprp-cactus/yprp-cactus.gbz -M -O pg -t 10- -x yprp-cactus/yprp-cactus.gbz
- use the Graph Burrows-Wheeler Transform generated by Cactus
- -M
- create a chunk for each path in the graph’s connected components, i.e. each chromosome
- -O pg
- the chunked graphs should be in the packed-graph format
- -t 10
- use 10 threads
BUG: Change the extension of the graph chunks from .vg to .pg:
for f in *.vg; do mv -- "$f" "${f%.vg}.pg"; doneMake a directory to store all the chromosome graphs:
mkdir chunks
mv *.pg chunks/Augment the packed-graph for chromosome 1 with the mapped reads (4min):
vg augment \
chunks/chunk_S288C#0#S288C.chrI.pg \
yprp-cactus.SK1.illumina.gam \
-s \
-m 4 \
-q 5 \
-Q 5 \
-A S288C.chrI.SK1.illumina.aug.gam \
-t 10 > S288C.chrI.SK1.illumina.aug.pg- -s
- the graph being augmented is a subgraph of the graph used to create the GAM
- -m 4
- filter breakpoints with coverage less than 4
- -q 5
- filter bases with quality less than 5
- -Q 5
- filter mappings with quality less than 5
- -A S288C.SK1.illumina.aug.gam
- output the read mappings relative to the augmented graph
- -t 10
- use 10 threads
- > S288C.chrI.SK1.illumina.aug.pg
- write the standard out (the augmented graph) into S288C.chrI.SK1.illumina.aug.pg
Compute read support for the augmented graph (<1min):
vg pack \
-x S288C.chrI.SK1.illumina.aug.pg \
-g S288C.chrI.SK1.illumina.aug.gam \
-Q 5 \
-s 5 \
-o S288C.chrI.SK1.illumina.aug.pack \
-t 10Compute snarls in the augmented graph structure (<1min):
vg snarls S288C.chrI.SK1.illumina.aug.pg -t 10 > S288C.chrI.SK1.illumina.aug.snarlsGenerate a VCF from the support (<1min)
vg call \
S288C.chrI.SK1.illumina.aug.pg \
-k S288C.chrI.SK1.illumina.aug.pack \
-r S288C.chrI.SK1.illumina.aug.snarls \
-t 10 > S288C.chrI.SK1.illumina.aug_calls.vcf10.7 BLAST the graph manually
Create a FASTA file containing the graph sequence (<1min):
gfatools gfa2fa yprp-cactus/yprp-cactus.gfa.gz > yprp-cactus.faBuild a BLAST database for the FASTA (1min):
makeblastdb -in yprp-cactus.fa -input_type fasta -dbtype nuclQuery the database for CUP1 and YHR054C (<1min):
blastn -outfmt 7 -db yprp-cactus.fa -query yprp/CUP1/cup1.yhr054c.fa > yprp-cactus.cup1.blast.tsv10.8 Viewing with Bandage
Cactus graphs are too complex so you should only view specific subgraphs with Bandage. Here we’ll look at a subgraph of chromosome 8 that contains the CUP1 gene.
List the paths in the chromosome 8 graph (<1min):
vg paths -L -x chunks/chunk_S288C#0#S288C.chrVIII.pg- -L
- List all paths in the graph
- -x chunks/chunk_S288C#0#S288C.chrVIII.pg
- The graph file to use
Compute snarls for the chromosome 8 graph (<1min):
vg snarls chunks/chunk_S288C#0#S288C.chrVIII.pg -t 10 > S288C.chrVIII.snarlsExtract the subgraph along the portion of the S288C path that contains the CUP1 gene (<1min):
vg chunk \
-x chunks/chunk_S288C#0#S288C.chrVIII.pg \
-S S288C.chrVIII.snarls \
-p S288C#0#S288C.chrVIII:210000-220000 \
-O gfa \
-t 10 > S288C.chrVIII.210000-220000.gfa- -x chunks/chunk_S288C#0#S288C.chrVIII.pg
- the graph to chunk
- -S S288C.chrVIII.snarls
- the snarls for the graph (can use -c for node distnace instead)
- -p S288C#0#S288C.chrVIII:210000-220000
- the path segment to extract
- -O gfa
- the output graph should be a GFA
- -t 10
- use 10 threads
- > S288C.chrVIII.210000-220000.gfa
- write the standard out (the graph chunk) into S288C.chrVIII.210000-220000.gfa
10.9 Pros and Cons PGGB
Pros:
- Building graphs is relatively fast
- Graphs represent multiple assmelbies
- Handles Giraffe indexing as part of graph building
Cons:
- Graphs are biased by the minigraph used to construct the graph
- Graphs are hard to visualize in Bandage
- Cactus pipeline is more opaque than vg construct
- Only “reference” path from minigraph is embedded in graph