Chapter 5 Introduction to Pangenomics
5.1 What is a “pangenome”?
The term “pangenome” was first coined by Sigaux et al. and was used to describe a public database containing an assessment of genome and transcriptome alterations in major types of tumors, tissues, and experimental models.
- Sigaux F. Génome du cancer ou de la construction des cartes d’identité moléculaire des tumeurs [Cancer genome or the development of molecular portraits of tumors]. Bull Acad Natl Med. 2000;184(7):1441-7; discussion 1448-9. French. PMID: 11261250.

The term was later revitalized by Tettelin et al. to describe a microbial genome by which genes were in the core (present in all strains) and which genes were dispensable (missing from one or more of the strains).

- Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., … & Fraser, C. M. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proceedings of the National Academy of Sciences, 102(39), 13950-13955.
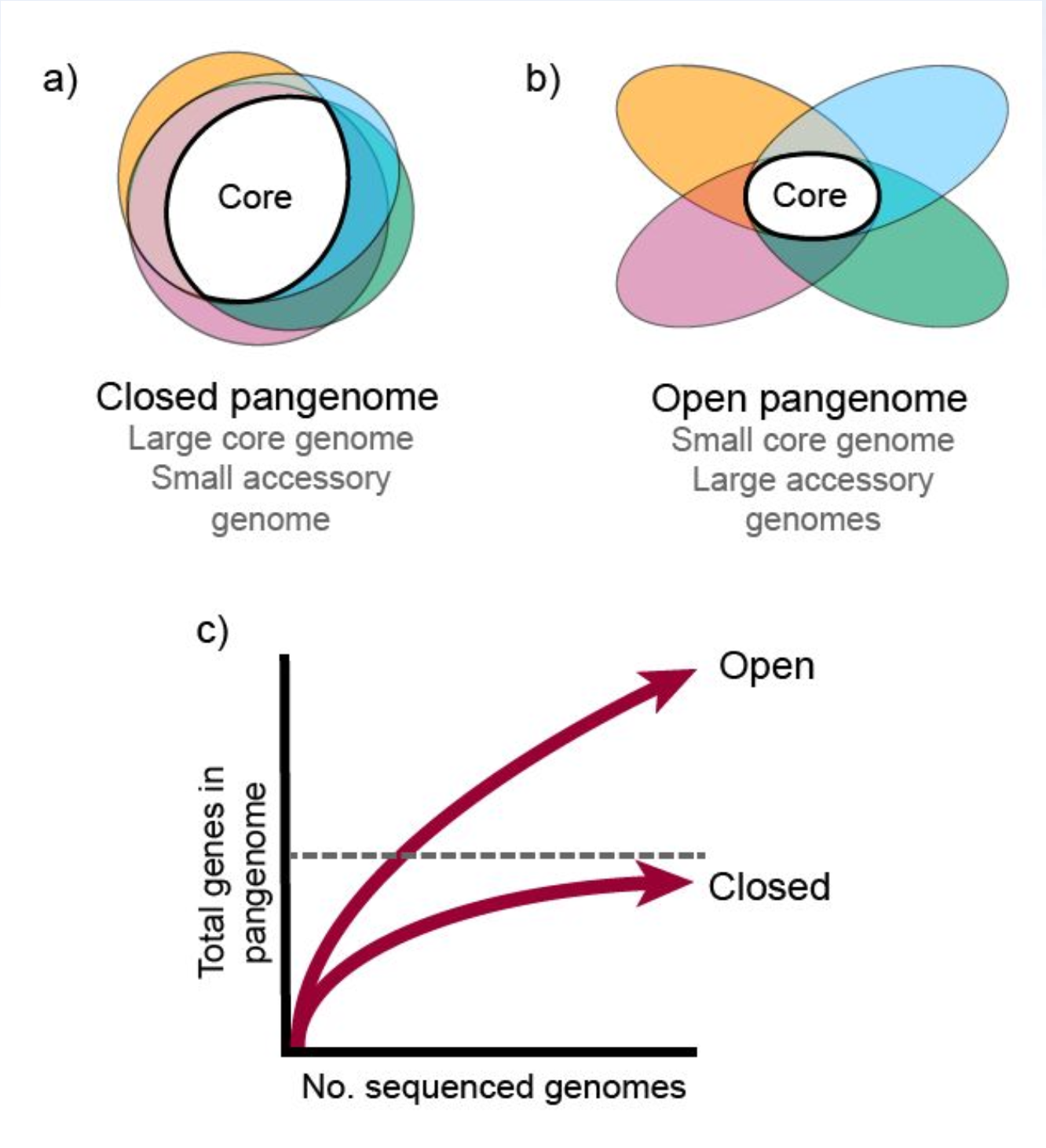
5.1.1 Open vs. Closed Genomes
5.1.2 Then vs. Now

- Low Cost
- High Quality Long Reads (HiFi)
- Many reference-quality assemblies per species

5.1.3 “Pangenome” Today
“Any collection of genomic sequences to be analyzed jointly or to be used as a reference. These sequences can be linked in a graph-like structure, or simply constitute sets of (aligned or unaligned) sequences.” – Computational Pangenomics Consortium
5.1.4 The Benefit of Pangenomes
- Removes reference bias
- May only represent one organism
- Could be a “mosaic”of individuals, i.e. doesn’t represent a coherent haplotype
- Allele bias
- Doesn’t include common variation
- Allow multiple assemblies to be analyzed simultaneously, i.e. efficiently
5.1.5 What are pangenomes good for?
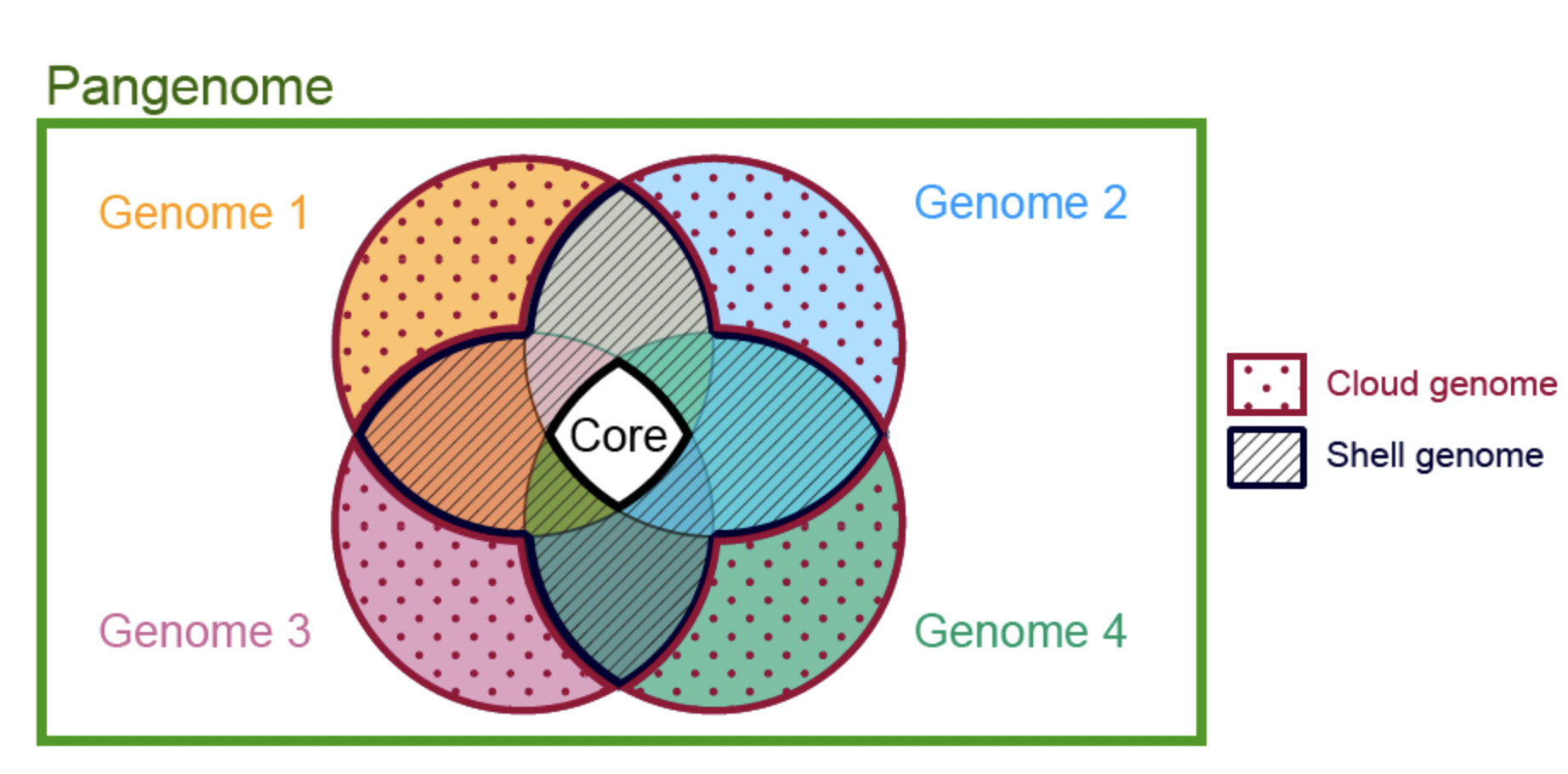
- Core vs dispensable genes:
- How big is the core?
- How big is the dispensable?
- How big is the pangenome?
- What traits are associated with the core/dispensable?
- Unbiased read mapping and variant calling
- More robust variation-trait association
- Visual exploration of genomic structure of population
5.2 Computational Pangenomics
“Questions about efficient data structures, algorithms and statistical methods to perform bioinformatic analyses of pan-genomes give rise to the discipline of ‘computational pan-genomics’.”
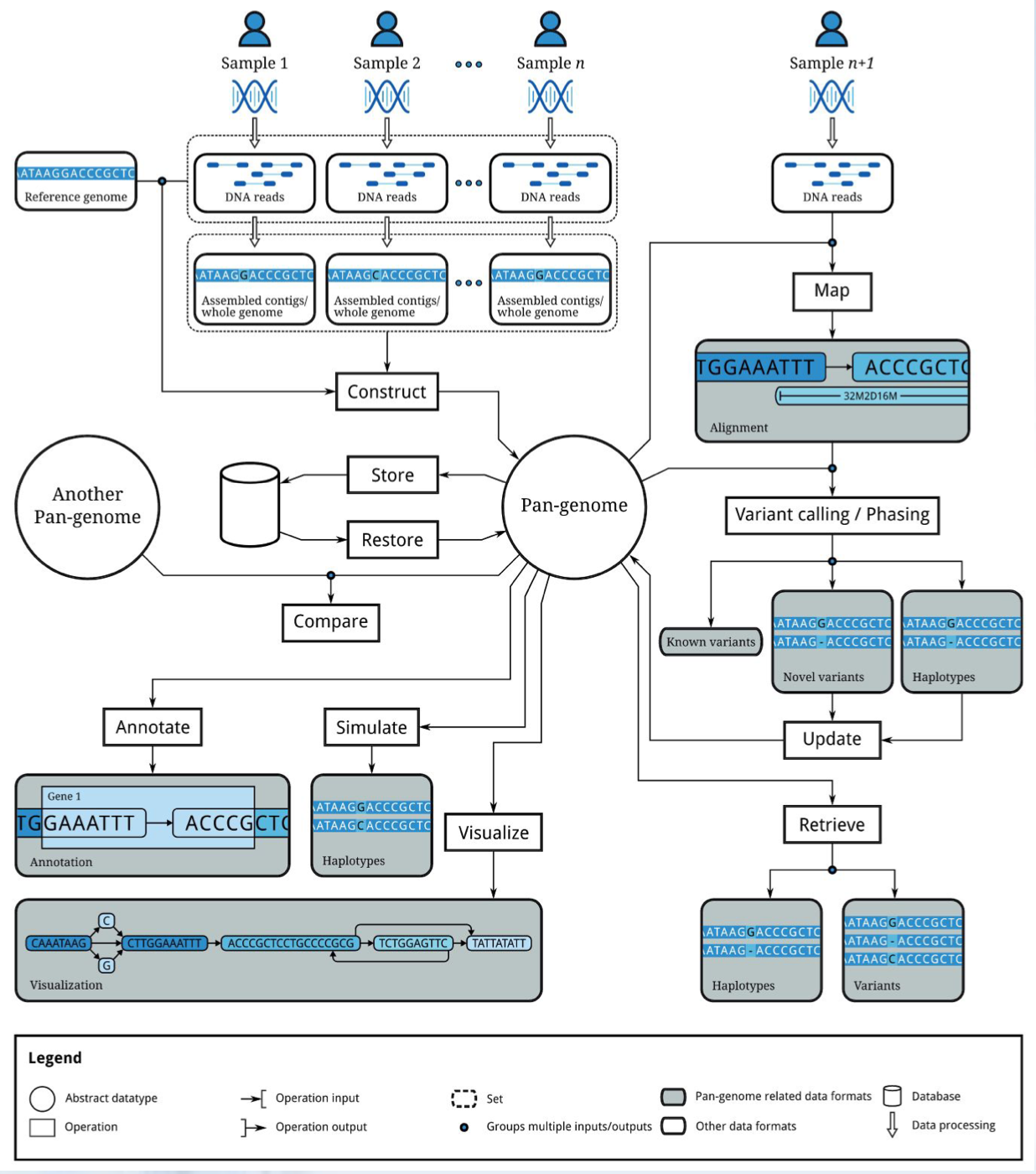
5.2.1 Pangenome Representations
- Gene sets
- Multiple sequence alignments
- K-mer sets
- Graphs
- De Bruijn graphs
- Haptotype graphs
- Variation graphs
5.2.2 Variation Graphs
- Variation forms bubbles Nodes represent sequences
- Chains of nodes represent contiguous sequence in one or more assemblies
- The sequences of nodes connected by an edge may overlap
- Graphs can be acyclic or cyclic
- Haplotypes are “threaded” through graph as paths
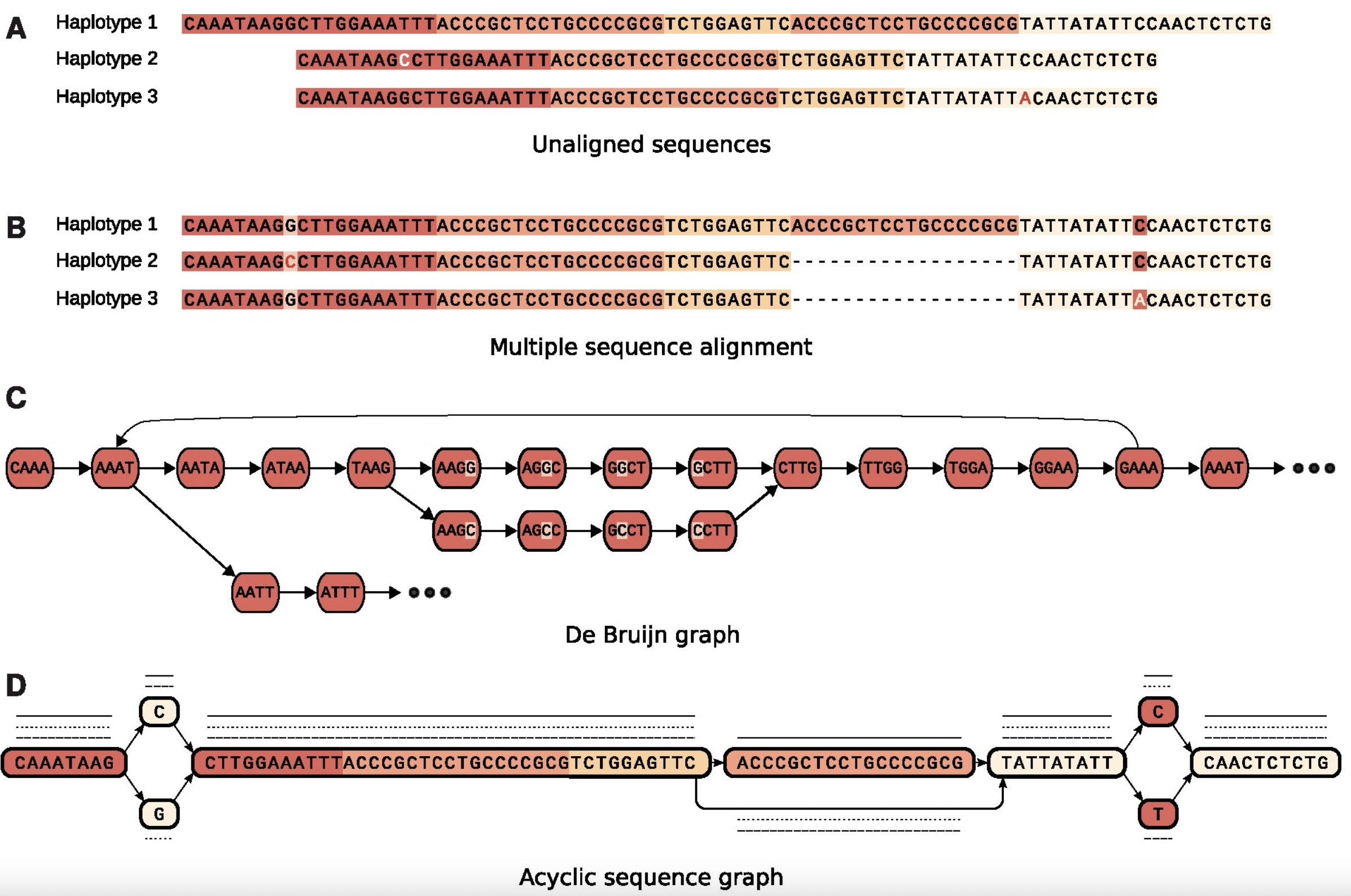
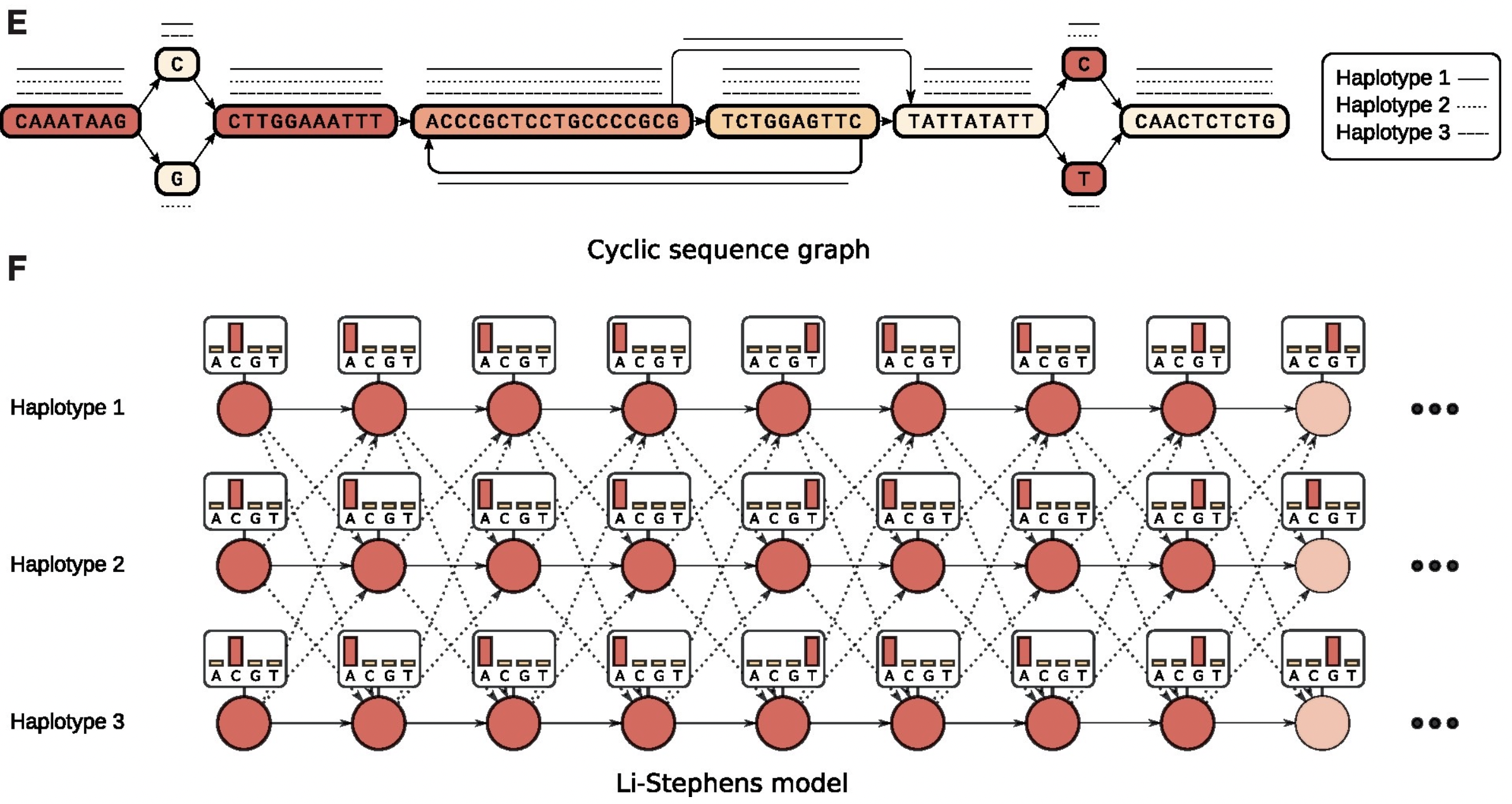
5.2.3 Types of Variation Graphs
- Reference Graph (vg)
- A reference with variants
- E.G. Human reference now includes VCF with common variation
- Reference Backbone; “iterative” (minigraph)
- Graph starts as reference and other sequences are layered on, i.e. variants can be relative to sequences other than the reference
- Reference-Free (Cactus and pggb)
- Graph is built using non-reference techniques, such as multiple sequence alignment
These are all methods used by the Human Pangenome Reference Consortium
5.2.4 Mapping Reads to Variation Graphs
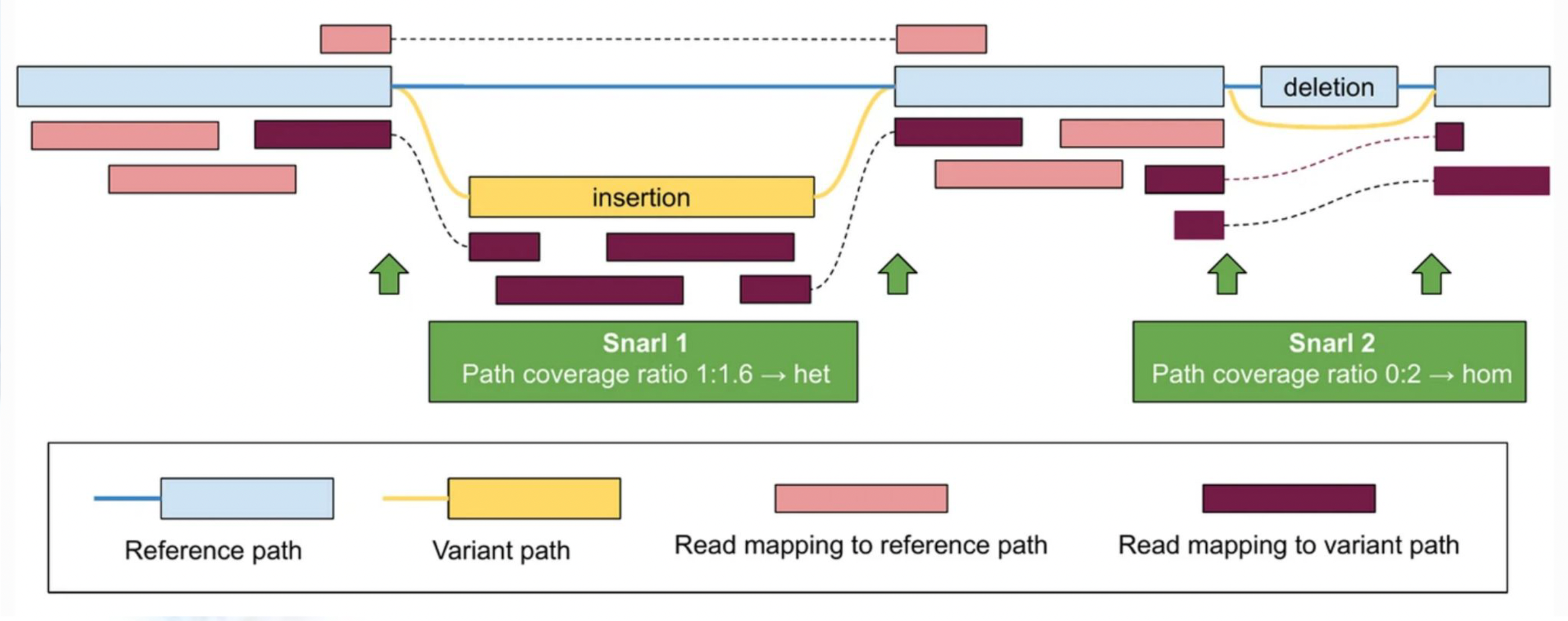
5.3 Pangenome Data Sets
- Human Reference + Variation VCF
- Zoonomia (200 mammalss) Project
- Maize NAM founder genomes
- Yeast Population Reference Panel (YPRP)
- Draft Human Pangenome Reference
5.3.1 Data/Yeast Genomes:
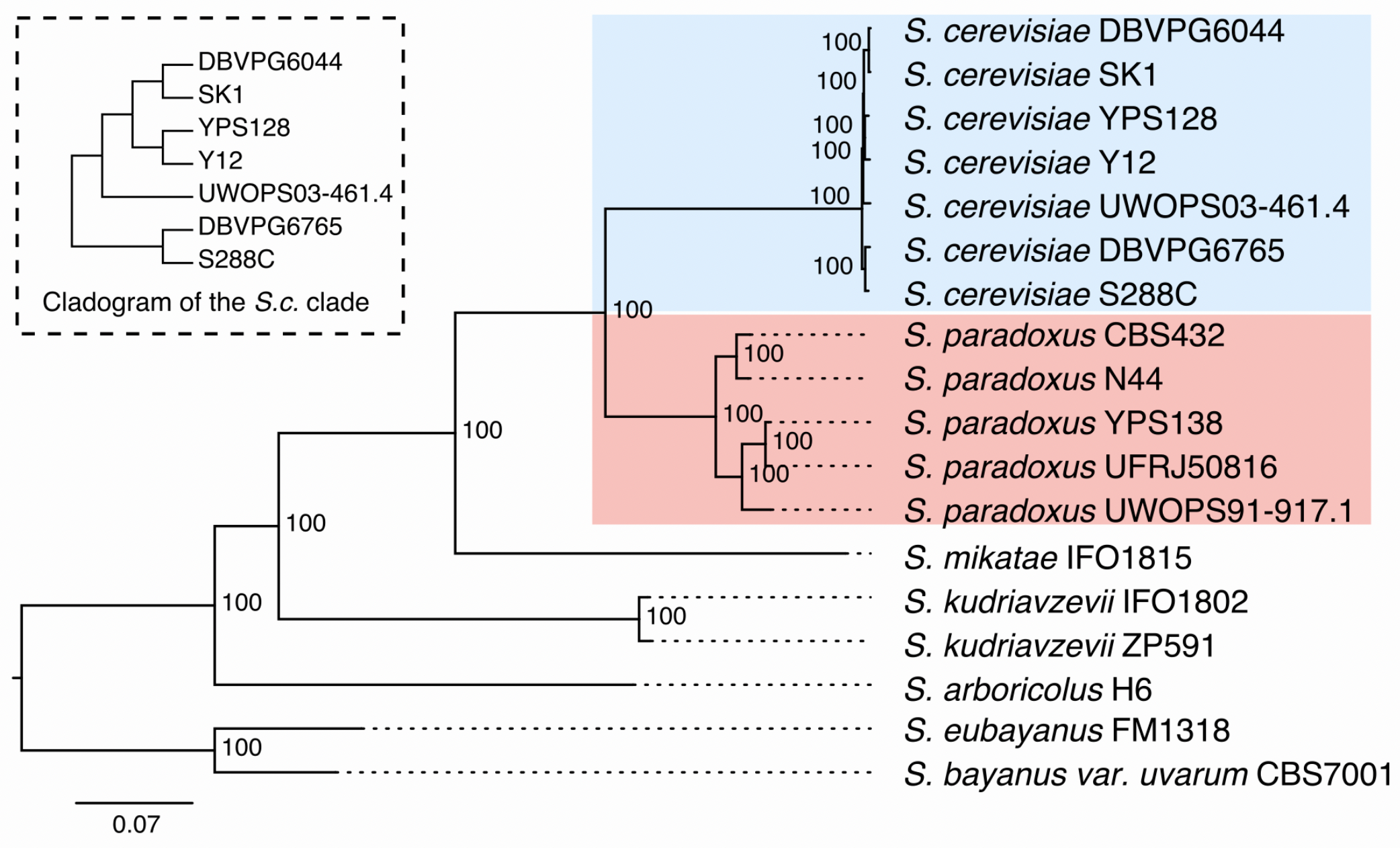
12 Mb
16 chromosomes
12 strains from Yeast Population Reference Panel (YPRP)
- 7 Saccharomyces cerevisiae (brewer’s yeast)
- Includes S288C reference
- 7 Saccharomyces cerevisiae (brewer’s yeast)
5 Saccharomyces paradoxus (wild yeast)
Software (LRSDAY)
5.3.2 Yeast Assemblies
- YPRP: 12 Yeast PacBio Assemblies (Chromosome level)
- ~100-200x PacBio sequencing reads
- HGAP + Quiver polishing
- ~200-500x Illumina (Pilon correction)
- Manual curation
- Annotation
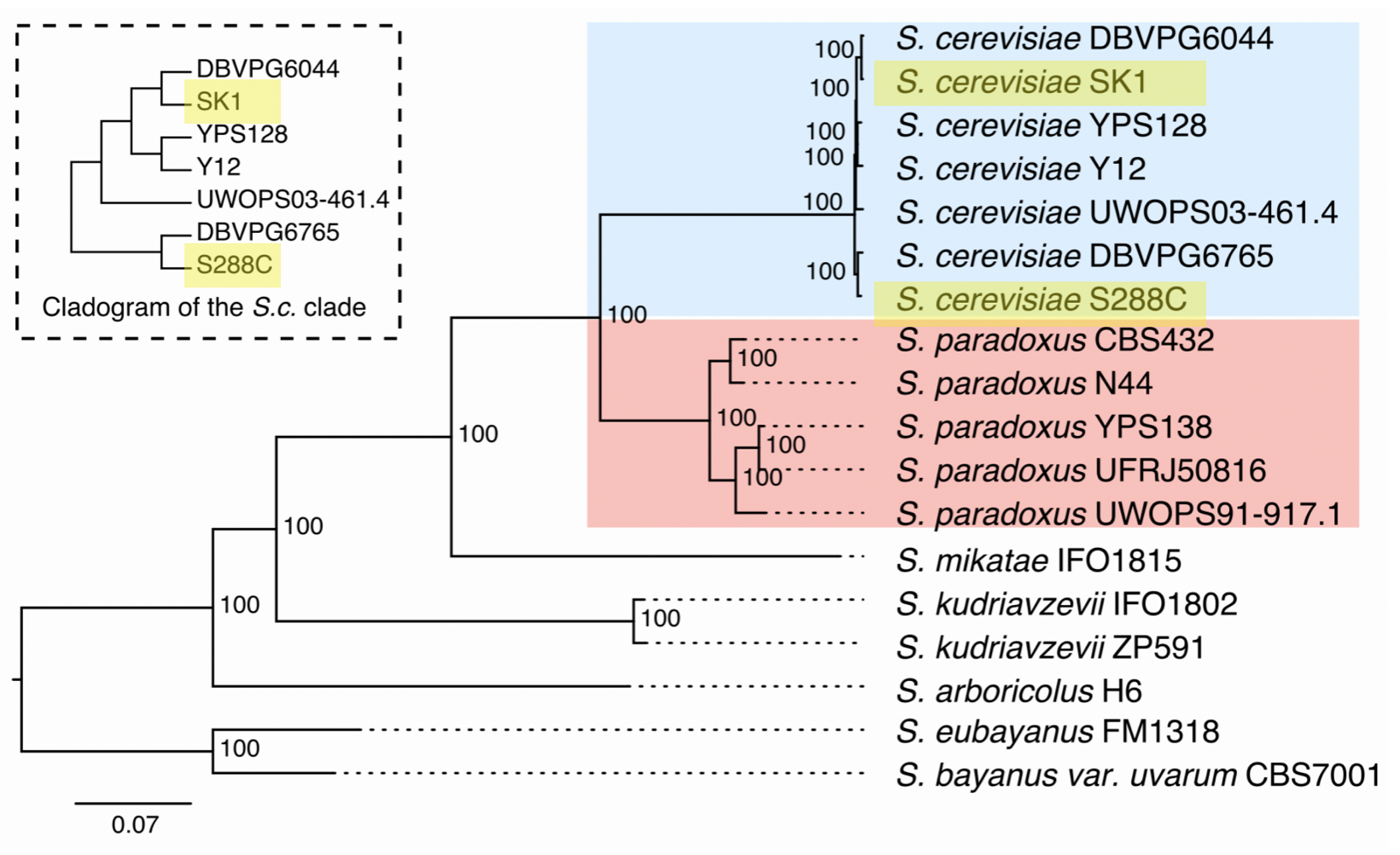
5.3.3 SK1 Illumina Reads
SK1 is the most distant from S288C
5.3.4 CUP1 Gene

5.3.5 We Changed the Names
- YPRP FASTA files only contain chromosome names
- We prefixed every chromosome with its assembly name and a “.” delineator
- e.g. S288C.chrVIII
- Pangenome Sequence Naming Specification