7 Seurat
Seurat is another R package for single cell analysis, developed by the Satija Lab. In this module, we will repeat many of the same analyses we did with SingleCellExperiment, while noting differences between them.
Important note: In this workshop, we use Seurat v5. If you find yourself working with a previous version of Seurat (v4 or earlier), the following shows how to adapt the workshop code to avoid incompatibilities.
| Seurat v3, v4 | Seurat v5 | |
|---|---|---|
Accessing counts
|
seurat[["RNA"]]@counts
|
seurat[["RNA"]]$counts
|
Accessing data
|
seurat[["RNA"]]@data
|
seurat[["RNA"]]$data
|
Initial value of data
|
Identical to counts
|
Does not exist until you normalize counts
|
Creating another assay, like CPM
|
CreateAssayObject()
|
CreateAssay5Object()
|
| Integration |
Requires JoinLayers() before looking at differential expression between integrated datasets
|
Seurat documentation for more details.
First we load the packages we will need.
library(Seurat)
library(ggplot2)
library(gridExtra)7.1 Build a fake Seurat object from scratch
First we generate the same small matrix of counts as we did for SingleCellExperiment. Note that instead of creating row and column names as metadata, we should attach them to the counts matrix.
set.seed(42)
num_genes <- 12
num_cells <- 8
raw_counts <- as.integer(rexp(num_genes*num_cells, rate = 0.5))
raw_counts <- matrix(raw_counts, nrow = num_genes, ncol = num_cells)
rownames(raw_counts) <- paste("Gene", 1:num_genes, sep = "-")
colnames(raw_counts) <- paste("Cell", 1:num_cells, sep = "-")And finally we create and inspect a Seurat object.
fake_seurat <- CreateSeuratObject(counts = raw_counts, project = "Fake Seurat")
fake_seurat## An object of class Seurat
## 12 features across 8 samples within 1 assay
## Active assay: RNA (12 features, 0 variable features)
## 1 layer present: countsThe default assay is called “RNA” and is accessible from fake_seurat@assays$RNA or just fake_seurat[["RNA"]]. This stores the raw counts in its counts slot, while its data slot is reserved for normalized data.
Assays(fake_seurat)## [1] "RNA"fake_seurat[["RNA"]]$counts## 12 x 8 sparse Matrix of class "dgCMatrix"
## Cell-1 Cell-2 Cell-3 Cell-4 Cell-5 Cell-6 Cell-7 Cell-8
## Gene-1 . . 2 . . . 1 2
## Gene-2 1 . 1 . 13 2 1 .
## Gene-3 . 2 2 . . . 2 .
## Gene-4 . . . 3 3 . 13 .
## Gene-5 . . . 1 2 . 2 .
## Gene-6 2 1 2 1 . . . 1
## Gene-7 . 2 1 . 1 3 1 5
## Gene-8 . . 6 2 . 1 2 3
## Gene-9 2 9 . 3 2 . 2 4
## Gene-10 1 1 . . 1 . . 2
## Gene-11 2 9 3 8 . 11 . 1
## Gene-12 4 . 1 2 16 . . 3fake_seurat[["RNA"]]$data## Warning: Layer 'data' is empty## 0 x 0 sparse Matrix of class "dgCMatrix"
## <0 x 0 matrix>Note that counts is a sparse matrix by default, and data has not yet been created.
7.1.1 Metadata
The meta.data slot is created with three columns by default.
head(fake_seurat@meta.data)## orig.ident nCount_RNA nFeature_RNA
## Cell-1 Fake Seurat 12 6
## Cell-2 Fake Seurat 24 6
## Cell-3 Fake Seurat 18 8
## Cell-4 Fake Seurat 20 7
## Cell-5 Fake Seurat 38 7
## Cell-6 Fake Seurat 17 4orig.ident is the project name we assigned this Seurat object, nCount_RNA is the total counts for each cell (like sum in SingleCellExperiment), and nFeature_RNA is the number of genes with nonzero counts for each cell (like detected).
7.1.2 Normalization and multiple assays
The Seurat normalization functions work slightly differently than in SingleCellExperiment, where multiple assays like logcounts, normcounts, and cpm naturally coexist. Instead, Seurat expects you to explicitly create a new assay for each (non-default) one, starting from the same counts. The transformed data are assigned to the new assay’s data slot, as in this example for counts per million. To compute CPM, use the relative counts (“RC”) method.
fake_seurat[["CPM"]] <- CreateAssay5Object(counts = fake_seurat[["RNA"]]$counts)
fake_seurat <- NormalizeData(fake_seurat, assay = "CPM", normalization.method = "RC", scale.factor = 1000000)## Normalizing layer: countsfake_seurat[["CPM"]]$data## 12 x 8 sparse Matrix of class "dgCMatrix"
## Cell-1 Cell-2 Cell-3 Cell-4 Cell-5 Cell-6 Cell-7
## Gene-1 . . 111111.11 . . . 41666.67
## Gene-2 83333.33 . 55555.56 . 342105.26 117647.06 41666.67
## Gene-3 . 83333.33 111111.11 . . . 83333.33
## Gene-4 . . . 150000 78947.37 . 541666.67
## Gene-5 . . . 50000 52631.58 . 83333.33
## Gene-6 166666.67 41666.67 111111.11 50000 . . .
## Gene-7 . 83333.33 55555.56 . 26315.79 176470.59 41666.67
## Gene-8 . . 333333.33 100000 . 58823.53 83333.33
## Gene-9 166666.67 375000.00 . 150000 52631.58 . 83333.33
## Gene-10 83333.33 41666.67 . . 26315.79 . .
## Gene-11 166666.67 375000.00 166666.67 400000 . 647058.82 .
## Gene-12 333333.33 . 55555.56 100000 421052.63 . .
## Cell-8
## Gene-1 95238.10
## Gene-2 .
## Gene-3 .
## Gene-4 .
## Gene-5 .
## Gene-6 47619.05
## Gene-7 238095.24
## Gene-8 142857.14
## Gene-9 190476.19
## Gene-10 95238.10
## Gene-11 47619.05
## Gene-12 142857.14To compute the log-normalized counts, use the (default) “LogNormalize” method. Note that the log-normalized counts use a different scale factor than in SingleCellExperiment, but this does not affect downstream computations.
fake_seurat <- NormalizeData(fake_seurat) # normalization.method = "LogNormalize"## Normalizing layer: countsfake_seurat[["RNA"]]$data## 12 x 8 sparse Matrix of class "dgCMatrix"
## Cell-1 Cell-2 Cell-3 Cell-4 Cell-5 Cell-6 Cell-7 Cell-8
## Gene-1 . . 7.014015 . . . 6.034684 6.860015
## Gene-2 6.726633 . 6.321767 . 8.137996 7.071124 6.034684 .
## Gene-3 . 6.726633 7.014015 . . . 6.726633 .
## Gene-4 . . . 7.313887 6.672632 . 8.597420 .
## Gene-5 . . . 6.216606 6.267800 . 6.726633 .
## Gene-6 7.419181 6.034684 7.014015 6.216606 . . . 6.167916
## Gene-7 . 6.726633 6.321767 . 5.576547 7.476306 6.034684 7.775676
## Gene-8 . . 8.112028 6.908755 . 6.378826 6.726633 7.265130
## Gene-9 7.419181 8.229778 . 7.313887 6.267800 . 6.726633 7.552637
## Gene-10 6.726633 6.034684 . . 5.576547 . . 6.860015
## Gene-11 7.419181 8.229778 7.419181 8.294300 . 8.775177 . 6.167916
## Gene-12 8.112028 . 6.321767 6.908755 8.345580 . . 7.265130Assays(fake_seurat)## [1] "RNA" "CPM"fake_seurat@active.assay## [1] "RNA"Now we have two assays, “RNA” and “CPM”. Both have the same counts but different data, and “RNA” is the active (default) one which (in this case) contains the log-normalized data.
7.2 Import a real one from our dataset
First we import the same data as before, using the Read10X() function.
my_counts <- Read10X("data/filtered_feature_bc_matrix/")
dim(my_counts)## [1] 34218 5780my_seurat <- CreateSeuratObject(counts = my_counts, project = "Arabidopsis")
my_seurat## An object of class Seurat
## 34218 features across 5780 samples within 1 assay
## Active assay: RNA (34218 features, 0 variable features)
## 1 layer present: counts# The following are equivalent, as you can see
# dim(my_seurat@assays$RNA$counts)
# dim(my_seurat[["RNA"]]$counts)
# dim(my_seurat[["RNA"]])
dim(my_seurat)## [1] 34218 5780dim(my_seurat@meta.data)## [1] 5780 3head(my_seurat@meta.data)## orig.ident nCount_RNA nFeature_RNA
## AAACCCACACGCGCAT-1 Arabidopsis 2285 1382
## AAACCCACAGAAGTTA-1 Arabidopsis 2609 1525
## AAACCCACAGACAAAT-1 Arabidopsis 4712 2606
## AAACCCACATTGACAC-1 Arabidopsis 6133 3434
## AAACCCAGTAGCTTTG-1 Arabidopsis 1243 998
## AAACCCAGTCCAGTTA-1 Arabidopsis 1095 914The metadata at this point are orig.ident (the project name), nCount_RNA (sum from my_SCE), and nFeature_RNA (detected from my_SCE).
We can also add the predetermined clusters.
df.clusters <- readRDS("data/clusters_5.rds")
df.clusters$cell_barcode <- substring(df.clusters$cell_barcode, 1, 18)
my_seurat@meta.data <- merge(my_seurat@meta.data, df.clusters, by.x = "row.names", by.y = "cell_barcode", all.x = TRUE)
# It converted row.names(my_seurat@meta.data) to a spurious column called Row.names,
# so clean up
row.names(my_seurat@meta.data) <- my_seurat@meta.data$Row.names
my_seurat@meta.data$Row.names <- NULL
head(my_seurat@meta.data)## orig.ident nCount_RNA nFeature_RNA cluster_id
## AAACCCACACGCGCAT-1 Arabidopsis 2285 1382 15
## AAACCCACAGAAGTTA-1 Arabidopsis 2609 1525 15
## AAACCCACAGACAAAT-1 Arabidopsis 4712 2606 4
## AAACCCACATTGACAC-1 Arabidopsis 6133 3434 14
## AAACCCAGTAGCTTTG-1 Arabidopsis 1243 998 17
## AAACCCAGTCCAGTTA-1 Arabidopsis 1095 914 9
## supercluster
## AAACCCACACGCGCAT-1 Endodermal cells
## AAACCCACAGAAGTTA-1 Endodermal cells
## AAACCCACAGACAAAT-1 Atrichoblasts
## AAACCCACATTGACAC-1 Endodermal cells
## AAACCCAGTAGCTTTG-1 Stele cells
## AAACCCAGTCCAGTTA-1 Meristematic cells# Assign display colors for the 21 clusters and 6 superclusters
# (close to those used in Farmer et al.)
cluster_colors <- c(
"cornflowerblue", "darkblue", "cyan",
"springgreen", "olivedrab", "chartreuse", "darkgreen",
"gray", "black", "saddlebrown",
"slateblue", "purple", "pink", "lightpink3", "hotpink", "darkred",
"lightsalmon", "orange", "tan", "firebrick1", "indianred"
)
supercluster_colors <- c("blue", "green", "black", "magenta", "red", "orange")7.2.1 Creating other metadata columns
Here we will create artificial metadata columns corresponding to the Arabidopsis thaliana gene name prefixes (AT1-AT5, ATC, ATM, and ENS). We use Seurat’s PercentageFeatureSet() function to compute the percentage of cell counts belonging to each category.
gene_prefix <- substring(rownames(my_seurat), 1, 3)
feature_categories <- sort(unique(gene_prefix))
for (fc in feature_categories) {
fc_name <- paste0("pct_", fc)
my_seurat[[fc_name]] <- PercentageFeatureSet(my_seurat, pattern = paste0("^", fc))
}
head(my_seurat@meta.data)## orig.ident nCount_RNA nFeature_RNA cluster_id
## AAACCCACACGCGCAT-1 Arabidopsis 2285 1382 15
## AAACCCACAGAAGTTA-1 Arabidopsis 2609 1525 15
## AAACCCACAGACAAAT-1 Arabidopsis 4712 2606 4
## AAACCCACATTGACAC-1 Arabidopsis 6133 3434 14
## AAACCCAGTAGCTTTG-1 Arabidopsis 1243 998 17
## AAACCCAGTCCAGTTA-1 Arabidopsis 1095 914 9
## supercluster pct_AT1 pct_AT2 pct_AT3 pct_AT4
## AAACCCACACGCGCAT-1 Endodermal cells 24.11379 16.10503 22.27571 17.41794
## AAACCCACAGAAGTTA-1 Endodermal cells 26.94519 15.82982 20.96589 14.25834
## AAACCCACAGACAAAT-1 Atrichoblasts 25.65789 16.02292 20.47963 16.08659
## AAACCCACATTGACAC-1 Endodermal cells 27.19713 15.32692 18.21295 16.24001
## AAACCCAGTAGCTTTG-1 Stele cells 24.29606 16.41191 21.31939 16.00965
## AAACCCAGTCCAGTTA-1 Meristematic cells 25.20548 13.78995 20.45662 15.89041
## pct_AT5 pct_ATC pct_ATM pct_ENS
## AAACCCACACGCGCAT-1 19.69365 0.04376368 0.35010941 0
## AAACCCACAGAAGTTA-1 21.08087 0.00000000 0.91989268 0
## AAACCCACAGACAAAT-1 21.73175 0.00000000 0.02122241 0
## AAACCCACATTGACAC-1 20.80548 0.35871515 1.85879667 0
## AAACCCAGTAGCTTTG-1 21.64119 0.08045052 0.24135157 0
## AAACCCAGTCCAGTTA-1 23.74429 0.18264840 0.73059361 0pdf("pdf/at1-5.pdf")
VlnPlot(my_seurat, features = paste0("pct_", "AT", 1:5), pt.size = 0, ncol = 5, fill.by = "feature", same.y.lims = TRUE)
dev.off()
pdf("pdf/atc-atm-ens.pdf")
VlnPlot(my_seurat, features = paste0("pct_", c("ATC", "ATM", "ENS")), pt.size = 0, ncol = 3, fill.by = "feature", same.y.lims = TRUE)
dev.off()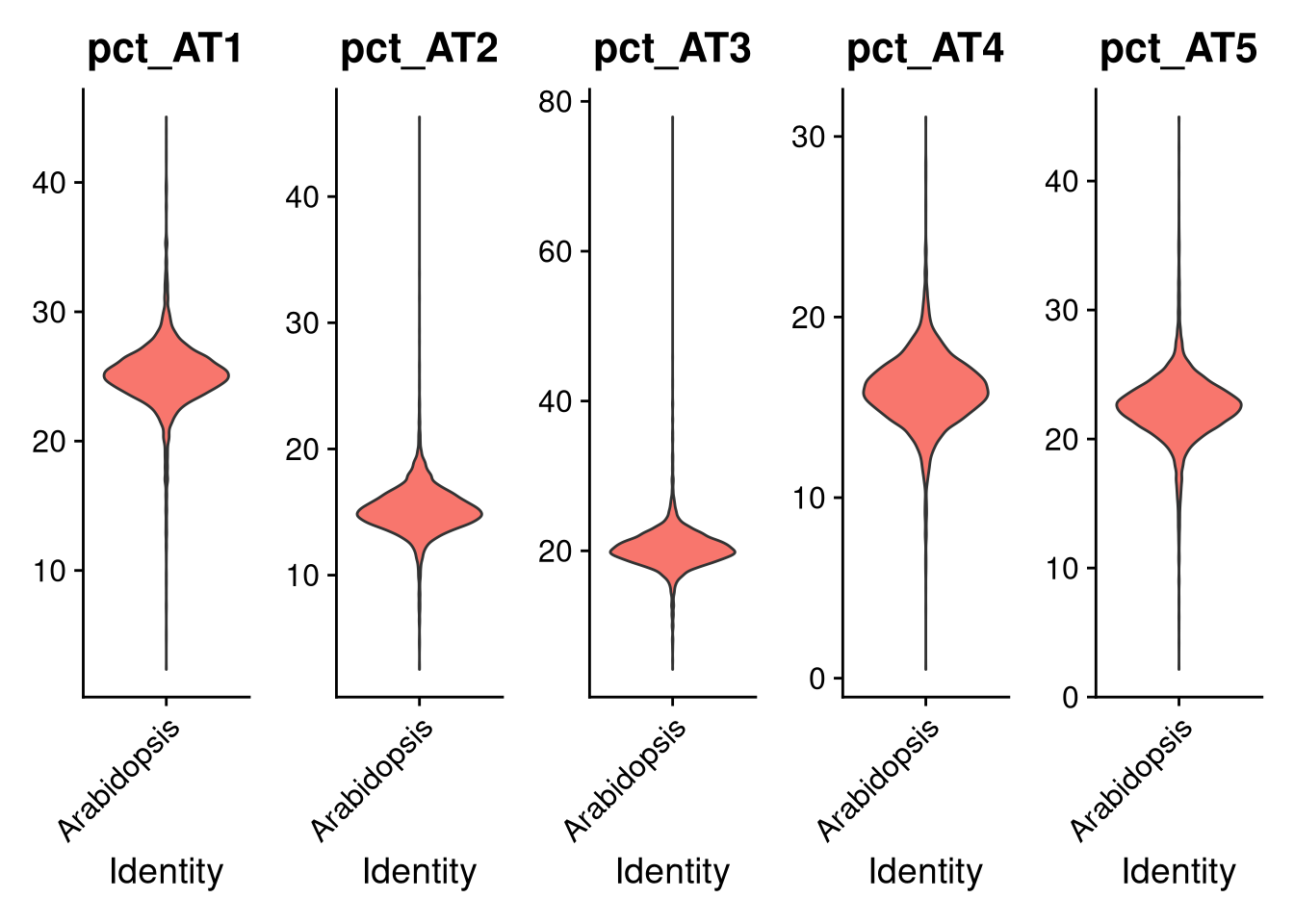
Figure 7.1: Violin plots of AT1-AT5 counts
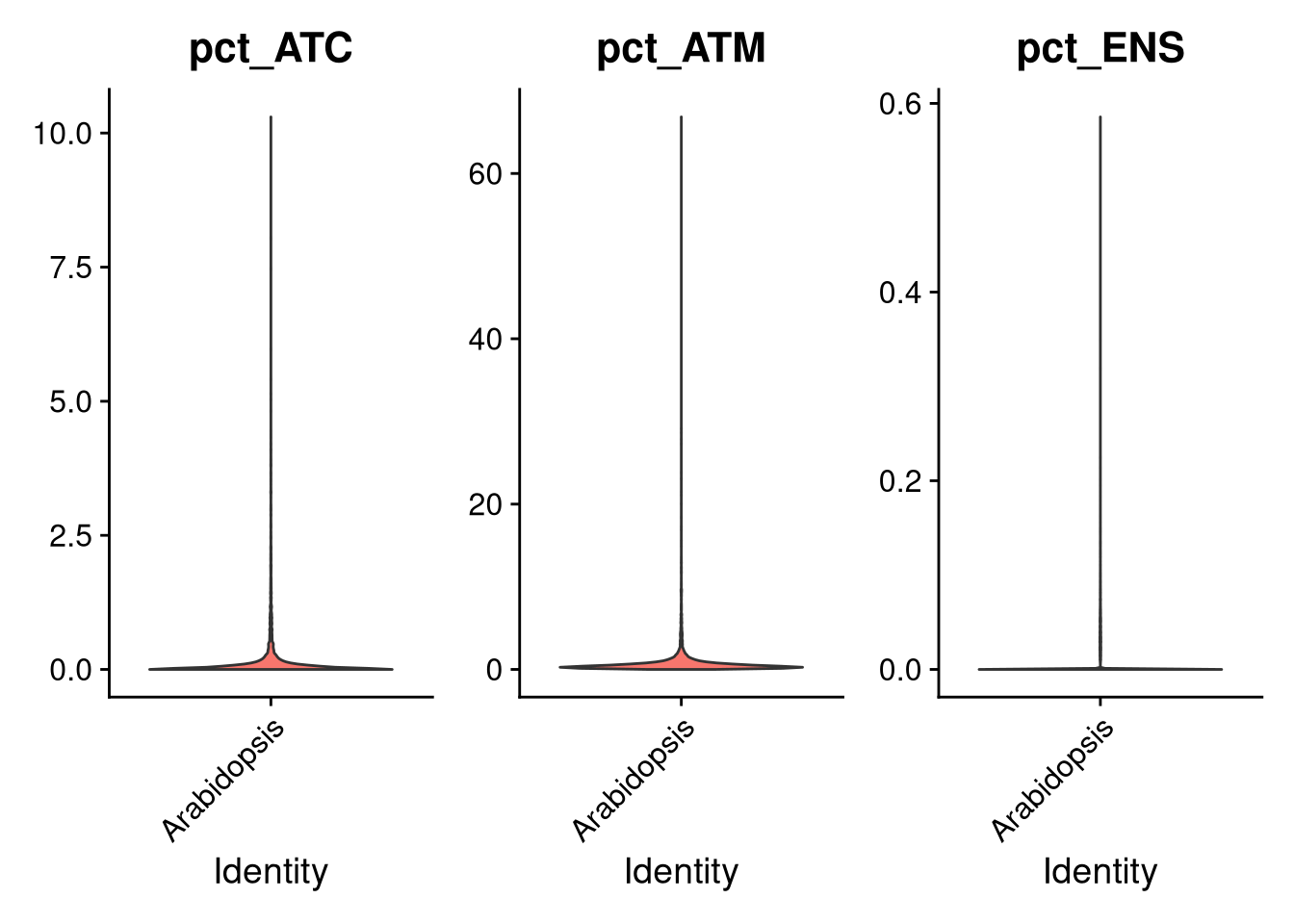
Figure 7.2: Violin plots of ATC, ATM, ENSRNA counts
The chloroplast, mitochondrial, and non-protein coding genes do not contribute much, so we are justified in eliminating them.
7.3 Quality control
We can decide which cells to discard using fixed thresholds. Before we filter out the discards, let’s illustrate FeatureScatter() which creates scatter plots for visualizing feature-feature relationships. It can be used for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
my_seurat$discard <- (my_seurat$nCount_RNA < 1000 | my_seurat$nFeature_RNA < 750 | is.na(my_seurat$cluster_id))pdf("pdf/seurat-fixed-threshold-qc.pdf")
FeatureScatter(my_seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA", group.by = "discard", cols = c("blue", "tan")) +
labs(color = "discard")
dev.off()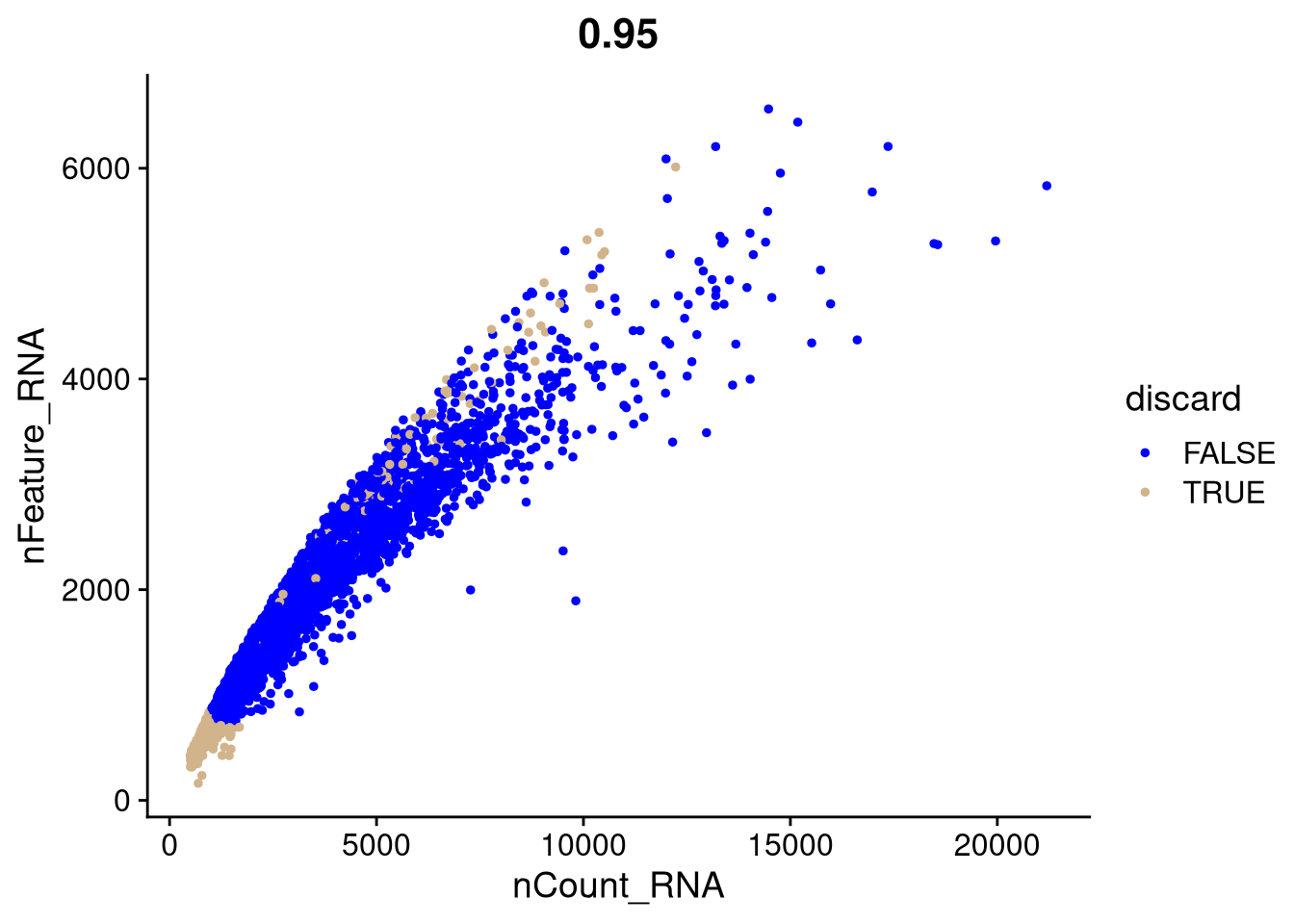
Figure 7.3: Cell counts v. number of expressed genes, showing outliers from fixed-threshold QC to discard
Now we can discard those cells and the undesired genes (“ATC”, “ATM”, “ENS”, and those with no cell counts).
genes_to_keep <- (rowSums(my_seurat[["RNA"]]$counts) > 0)
genes_to_keep <- genes_to_keep & gene_prefix %in% paste0("AT", 1:5)
my_seurat <- my_seurat[genes_to_keep, !my_seurat$discard]
dim(my_seurat)## [1] 23848 51517.4 Plots
VlnPlot() shows the variation in individual features. By default, this overlays the violin plot with the jittered count values, so it is difficult to see. To remove them, we add the argument pt.size = 0.
pdf("pdf/nCount-nFeature-by-cluster.pdf")
plot1 <- VlnPlot(my_seurat, features = "nCount_RNA", cols = cluster_colors, pt.size = 0, group.by = "cluster_id") +
xlab("cluster_id") +
NoLegend()
plot2 <- VlnPlot(my_seurat, features = "nFeature_RNA", cols = cluster_colors, pt.size = 0, group.by = "cluster_id") +
xlab("cluster_id") +
NoLegend()
grid.arrange(plot1, plot2, ncol = 1)
dev.off()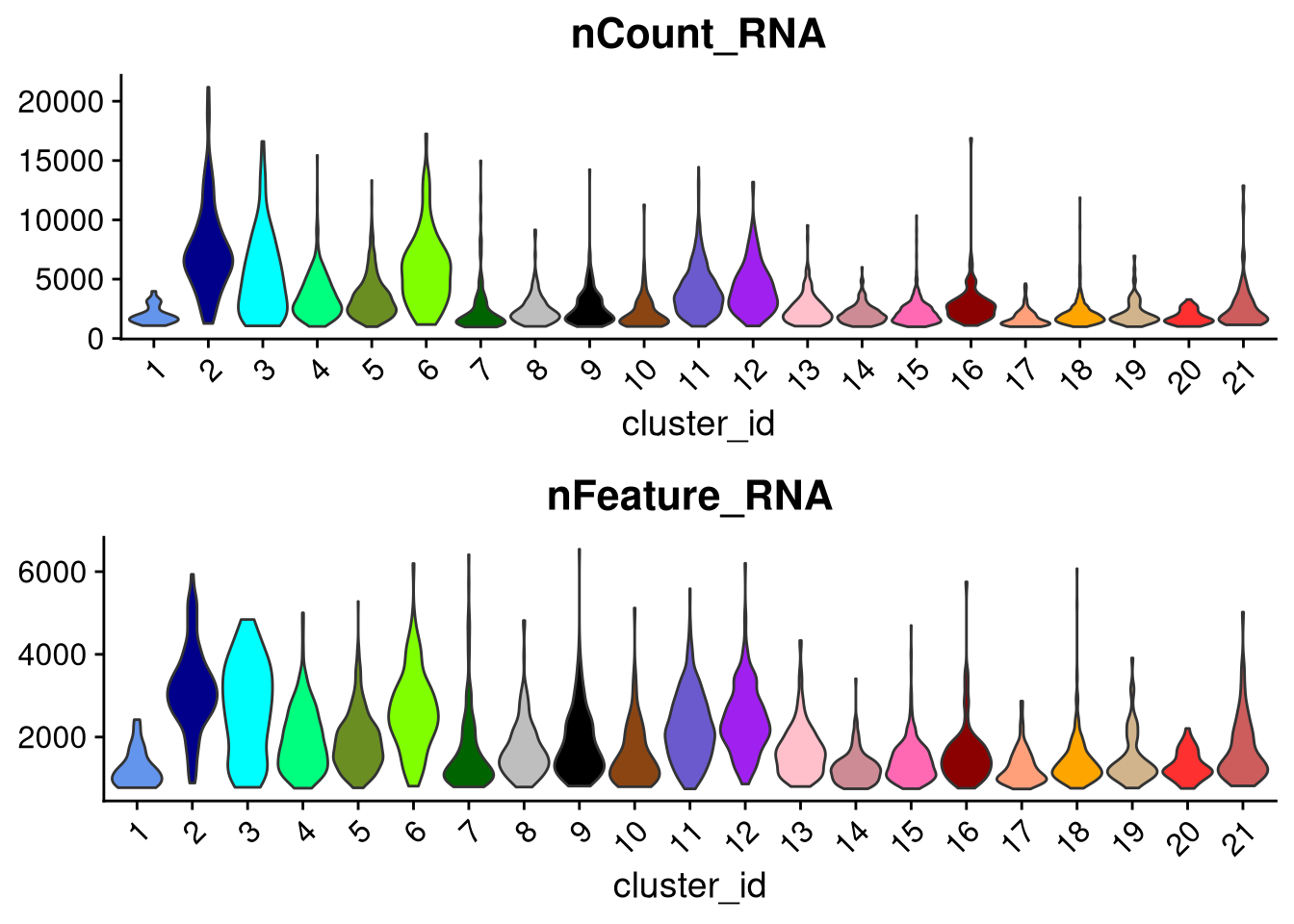
Figure 7.4: Cell counts and number of expressed genes, by cluster
The group.by argument determines which categories of cell to give their own violin plot. For example, to group by supercluster instead of cluster_id,
pdf("pdf/nCount-nFeature-by-supercluster.pdf")
plot1 <- VlnPlot(my_seurat, features = "nCount_RNA", cols = supercluster_colors, pt.size = 0, group.by = "supercluster") +
xlab("supercluster") +
NoLegend()
plot2 <- VlnPlot(my_seurat, features = "nFeature_RNA", cols = supercluster_colors, pt.size = 0, group.by = "supercluster") +
xlab("supercluster") +
NoLegend()
grid.arrange(plot1, plot2, ncol = 2)
dev.off()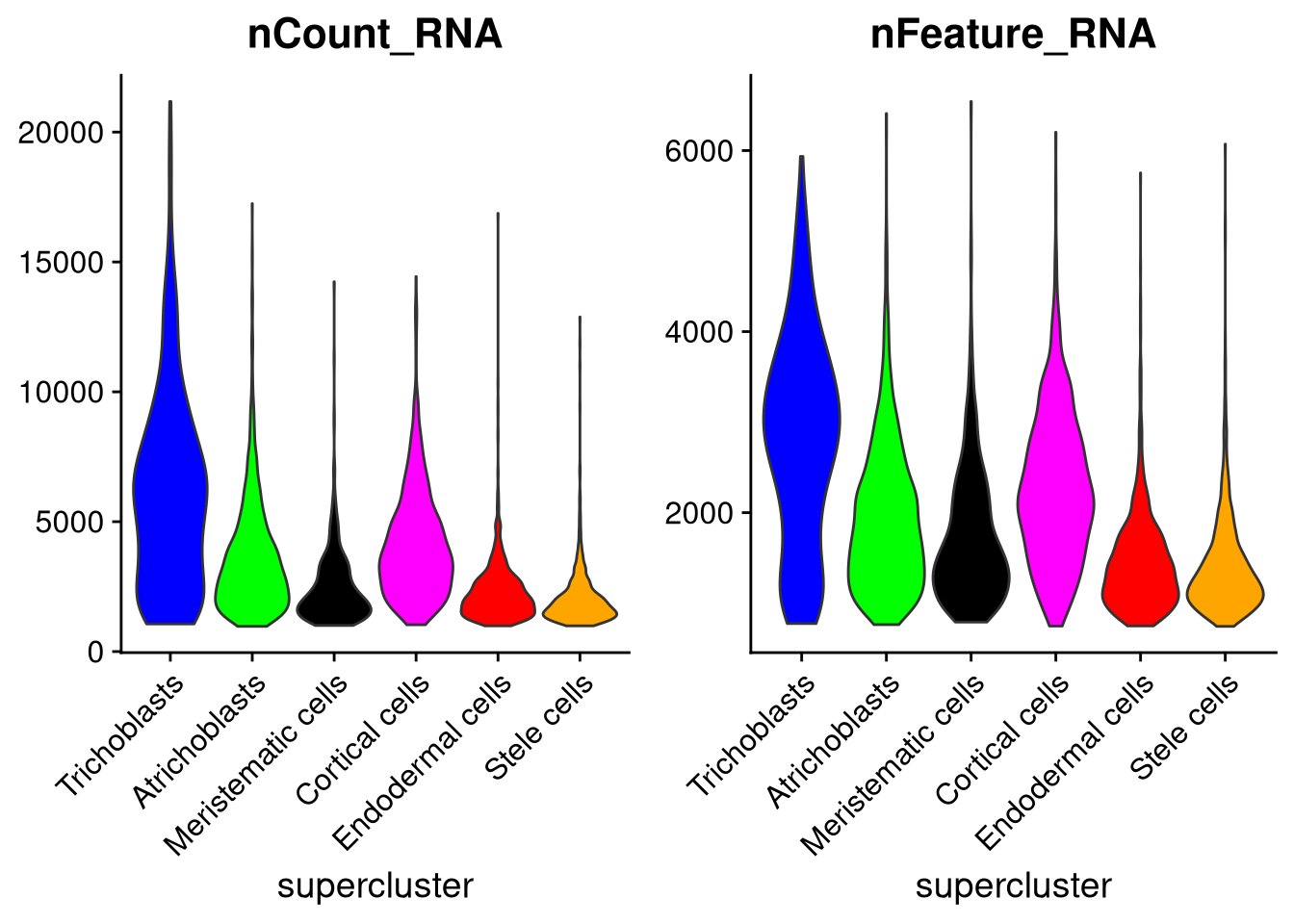
Figure 7.5: Cell counts and number of expressed genes, by supercluster
We can also visualize the same data as a ridgeline plot.
pdf("pdf/nCount-nFeature-ridgeplots.pdf")
plot1 <- RidgePlot(my_seurat, features = "nCount_RNA", cols = supercluster_colors, group.by = "supercluster") +
ylab(NULL) +
NoLegend()
plot2 <- RidgePlot(my_seurat, features = "nFeature_RNA", cols = supercluster_colors, group.by = "supercluster") +
ylab(NULL) +
NoLegend()
grid.arrange(plot1, plot2, ncol = 1)
dev.off()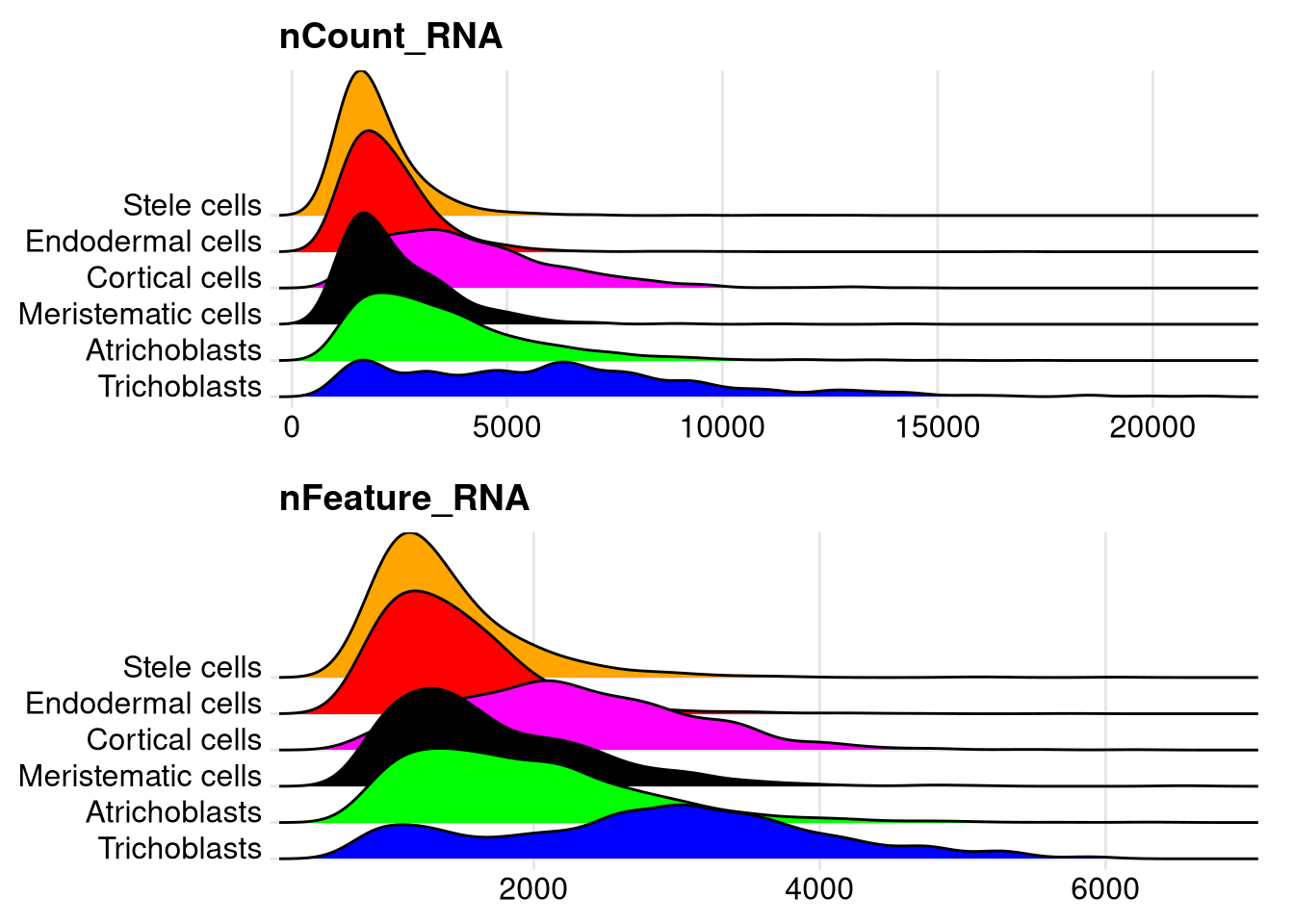
Figure 7.6: Ridgeline plots of cell counts and number of expressed genes, by supercluster
To combine all data into a single category, group by orig.ident.
7.5 Normalization
Again, we can log-normalize our data just by running NormalizeData().
my_seurat <- NormalizeData(my_seurat)## Normalizing layer: countsmy_seurat[["RNA"]]$counts[1:6, 1:3]## 6 x 3 sparse Matrix of class "dgCMatrix"
## AAACCCACACGCGCAT-1 AAACCCACAGAAGTTA-1 AAACCCACAGACAAAT-1
## AT1G01010 . 1 .
## AT1G01020 . . 1
## AT1G01030 . . .
## AT1G01040 . . 2
## AT1G03993 . . .
## AT1G01050 1 . .my_seurat[["RNA"]]$data[1:6, 1:3]## 6 x 3 sparse Matrix of class "dgCMatrix"
## AAACCCACACGCGCAT-1 AAACCCACAGAAGTTA-1 AAACCCACAGACAAAT-1
## AT1G01010 . 1.58278 .
## AT1G01020 . . 1.138695
## AT1G01030 . . .
## AT1G01040 . . 1.657348
## AT1G03993 . . .
## AT1G01050 1.685227 . .7.6 Identification of highly variable features
Another method of quality control is to determine the most variable genes and use these for dimensionality reduction. Here we use FindVariableFeatures() to identify the 2000 most variable genes (with the top 10 labeled), and VariableFeaturePlot() to visually compare them to the less variable ones.
my_seurat <- FindVariableFeatures(my_seurat) # nfeatures = 2000 by default, unlike 500 as in SingleCellExperiment## Finding variable features for layer countstop_10_genes <- head(VariableFeatures(my_seurat), 10)pdf("pdf/variable-features.pdf")
vf_plot <- VariableFeaturePlot(my_seurat, cols = c("darkgray", "blue"))
LabelPoints(plot = vf_plot, points = top_10_genes, repel = TRUE, xnudge = 0, ynudge = 0)
dev.off()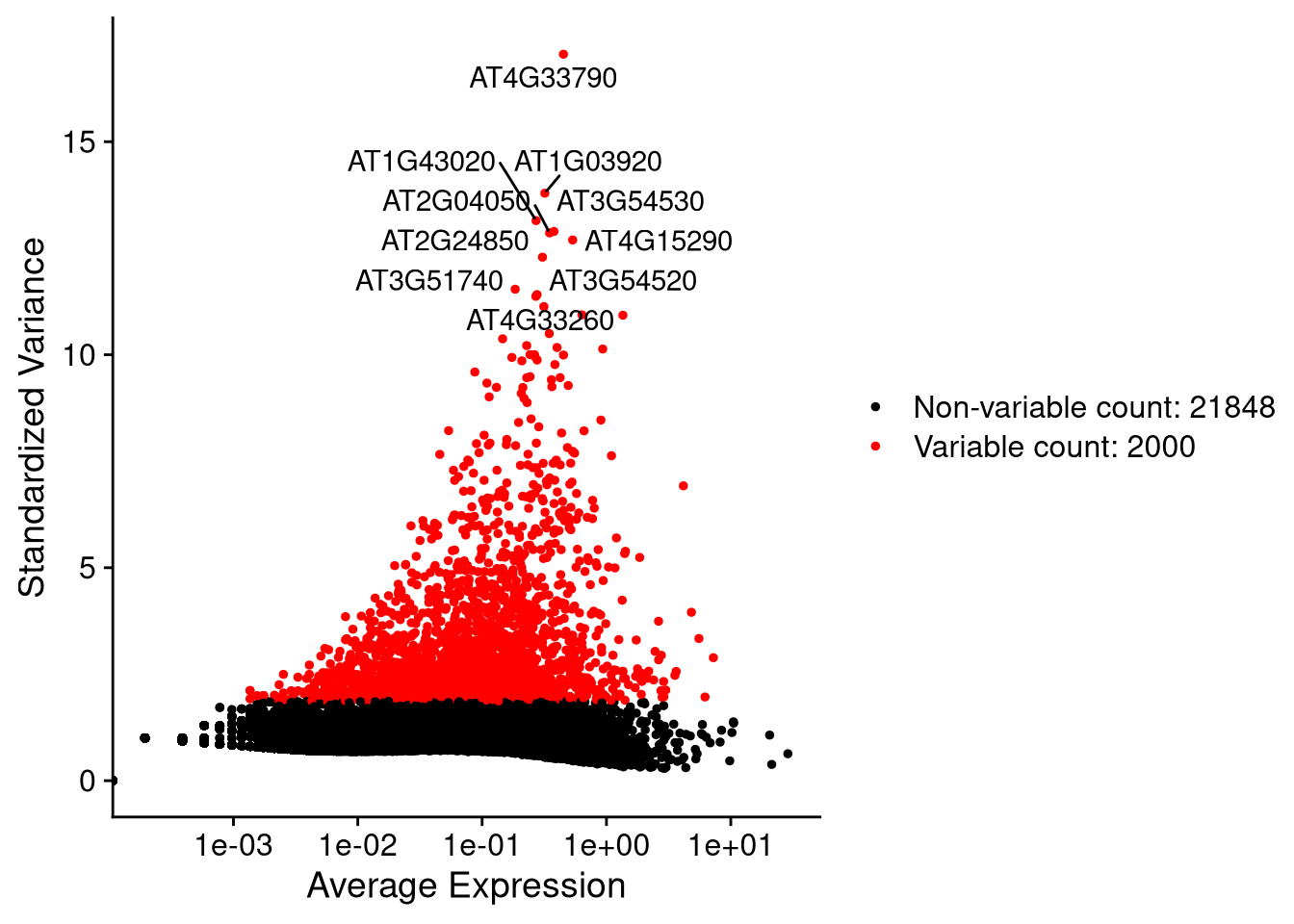
Figure 7.7: Variable feature plot
7.7 Dimensionality reduction
7.7.1 PCA
In Seurat, before computing principal components we must scale the (log-normalized) data by subtracting the mean and dividing by the standard deviation of each row. The features argument allows your choice of genes to scale, but the default is to only scale the rows corresponding to the most variable genes.
my_seurat <- ScaleData(my_seurat, verbose = FALSE)
my_seurat <- RunPCA(my_seurat, verbose = FALSE)We can view the individual PC values for each cell as follows.
# Just the first 6 rows and 5 columns
my_seurat[["pca"]]@cell.embeddings[1:6, 1:5]## PC_1 PC_2 PC_3 PC_4 PC_5
## AAACCCACACGCGCAT-1 -3.5730765 -0.720932 1.5214319 -3.1399856 -4.6600356
## AAACCCACAGAAGTTA-1 -4.6125194 -21.638604 -5.1986060 -0.9866993 -3.4633961
## AAACCCACAGACAAAT-1 1.8597980 2.741490 1.7616183 10.2241086 -1.9462996
## AAACCCACATTGACAC-1 -0.4922986 -8.080446 -0.8845399 0.8801926 -0.2192683
## AAACCCAGTAGCTTTG-1 -3.5552763 1.851916 4.3438592 -3.5047218 2.2177276
## AAACCCAGTCCAGTTA-1 -4.0690844 3.278787 -0.1907693 -1.4210729 3.8602152The VizDimLoadings() function shows which genes are most strongly associated with each principal component.
pdf("pdf/vizdimloadings.pdf")
VizDimLoadings(my_seurat, dims = 1:4, nfeatures = 10, reduction = "pca")
dev.off()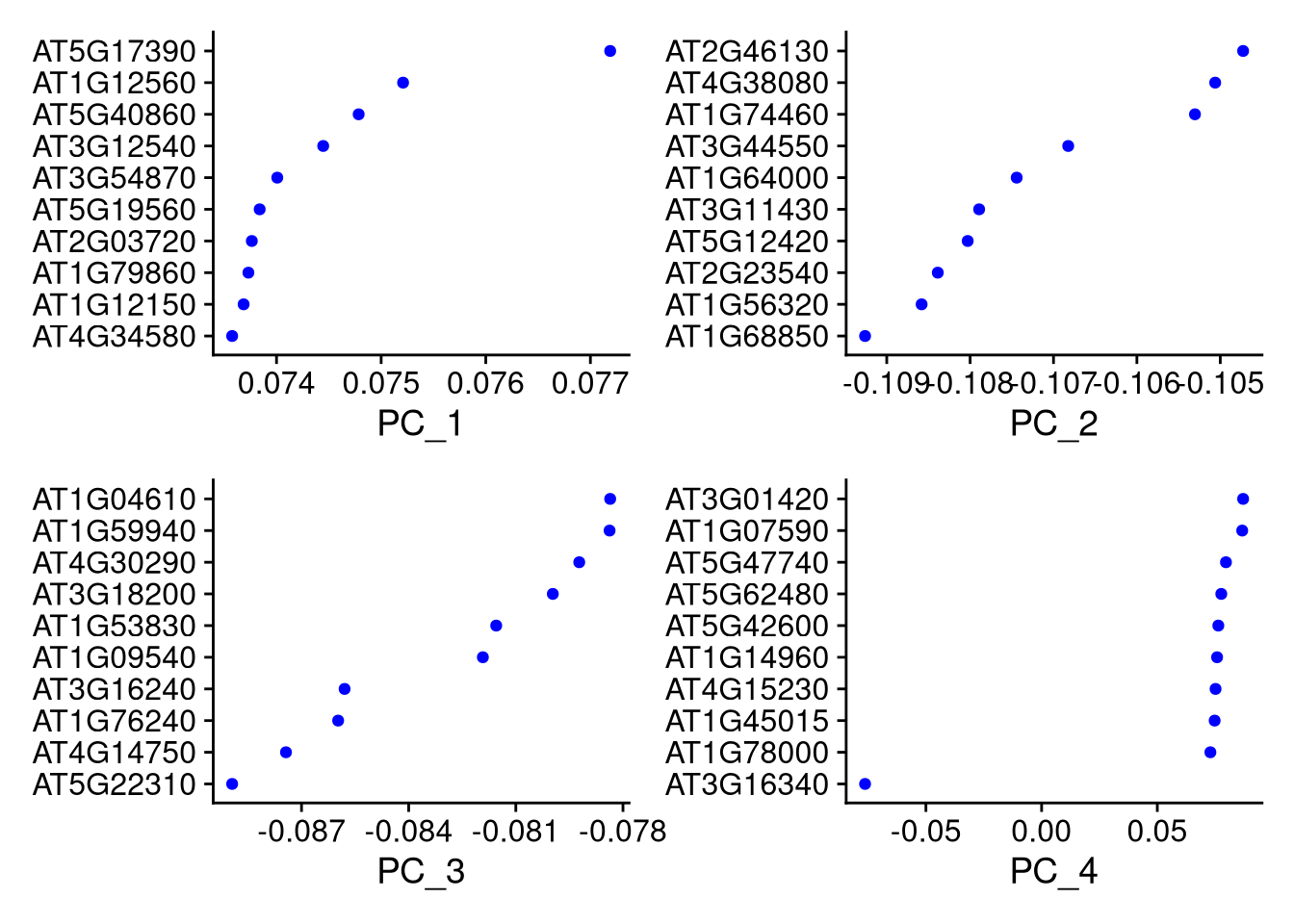
Figure 7.8: Dimensional loadings plot for the first 4 principal components
DimHeatmap() sorts both genes and cells by principal component values. Genes with the most negative PC values are on top, and those with the most positive values are on the bottom, while cells are arranged from left to right. In this default color scheme, magenta represents positive correlations and yellow represents negative ones, with no correlation in black.
pdf("pdf/dimheatmaps.pdf")
DimHeatmap(my_seurat, dims = 1:4, nfeatures = 10, ncol = 2, cells = 500)
dev.off()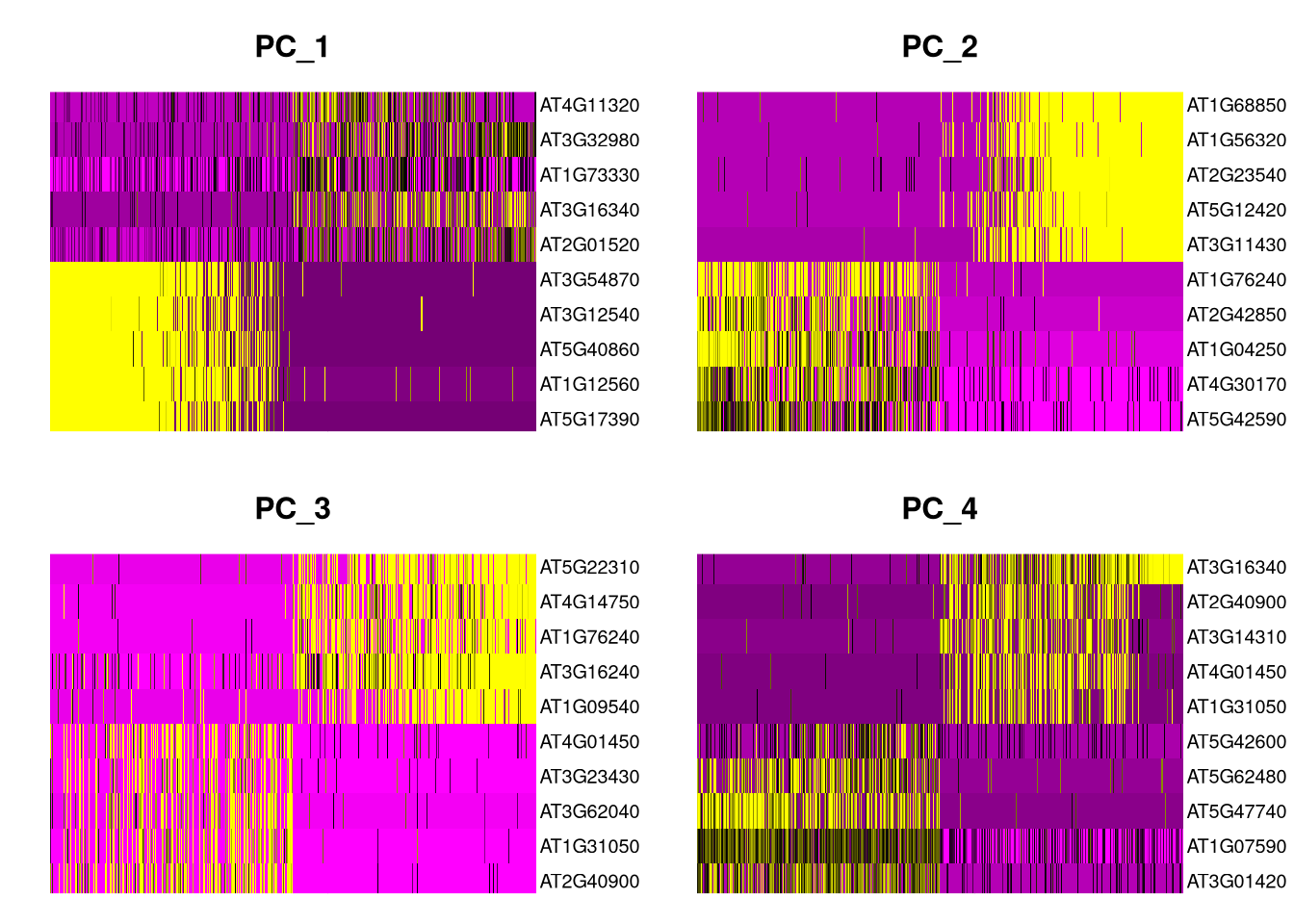
Figure 7.9: Heatmaps for the first 4 principal components, top 10 genes x 500 cells
VizDimLoadings(). Do they agree?
Click for answer
Yes, remember to count positive-valued genes from the bottom and negative-valued genes from the top of each heatmap.The ElbowPlot() function shows the amount of variance in each principal component, giving us a sense of how many PCs we should retain in subsequent computations.
pdf("pdf/elbow-plot-seurat.pdf")
ElbowPlot(my_seurat, ndims = 50)
dev.off()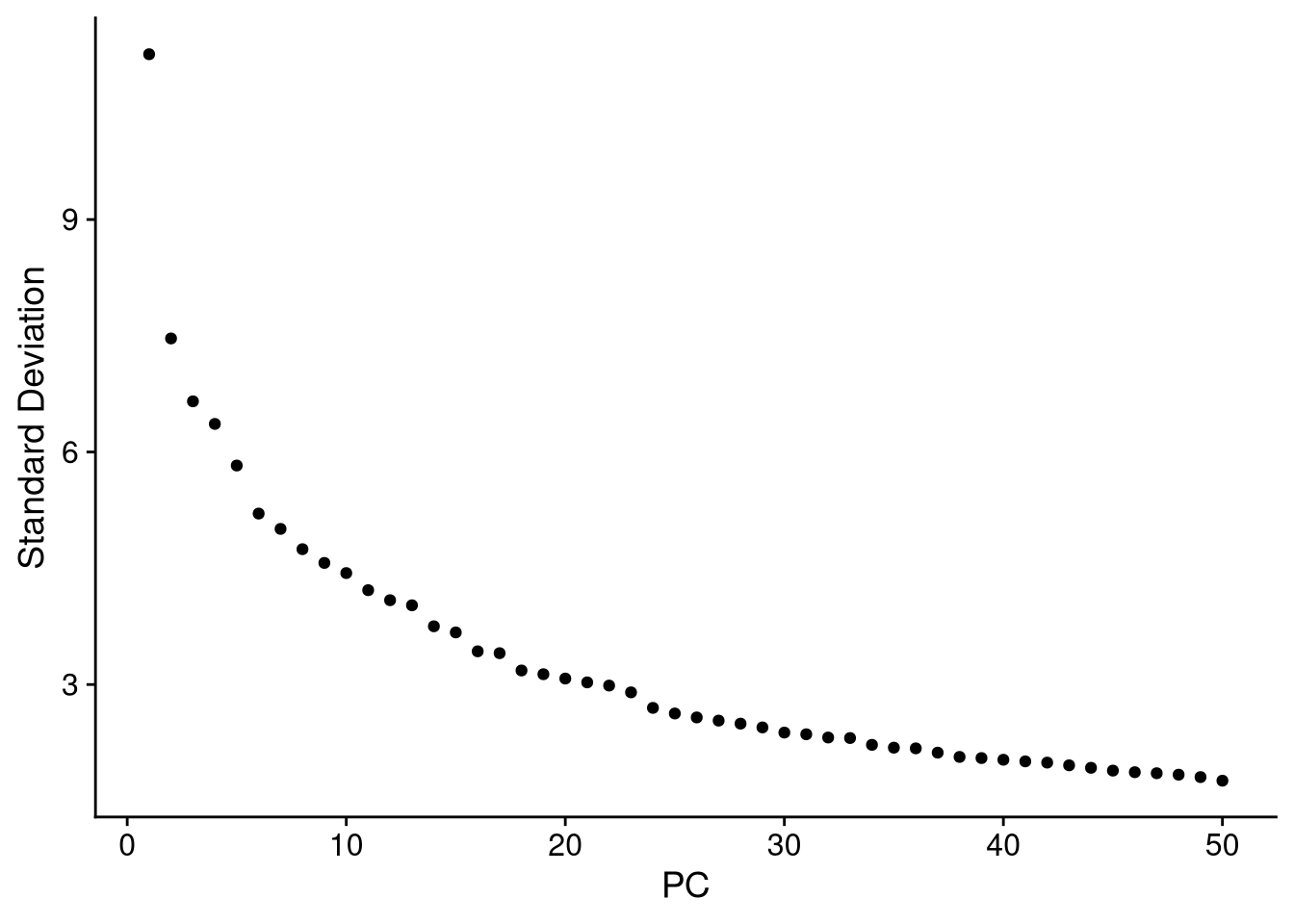
Figure 7.10: Elbow plot for 50 PCs
It looks like we would not gain much by retaining more than 20 PCs in our dimensionality reduction computations.
Finally, we can create our dimensionality reduction plots with DimPlot().
pdf("pdf/pca-by-cluster-seurat.pdf")
DimPlot(my_seurat, reduction = "pca", cols = cluster_colors, group.by = "cluster_id", label.size = 4, repel = TRUE, label = TRUE)
dev.off()
pdf("pdf/pca-by-supercluster-seurat.pdf")
DimPlot(my_seurat, reduction = "pca", cols = supercluster_colors, group.by = "supercluster", label.size = 4, repel = TRUE, label = TRUE) + NoLegend()
dev.off()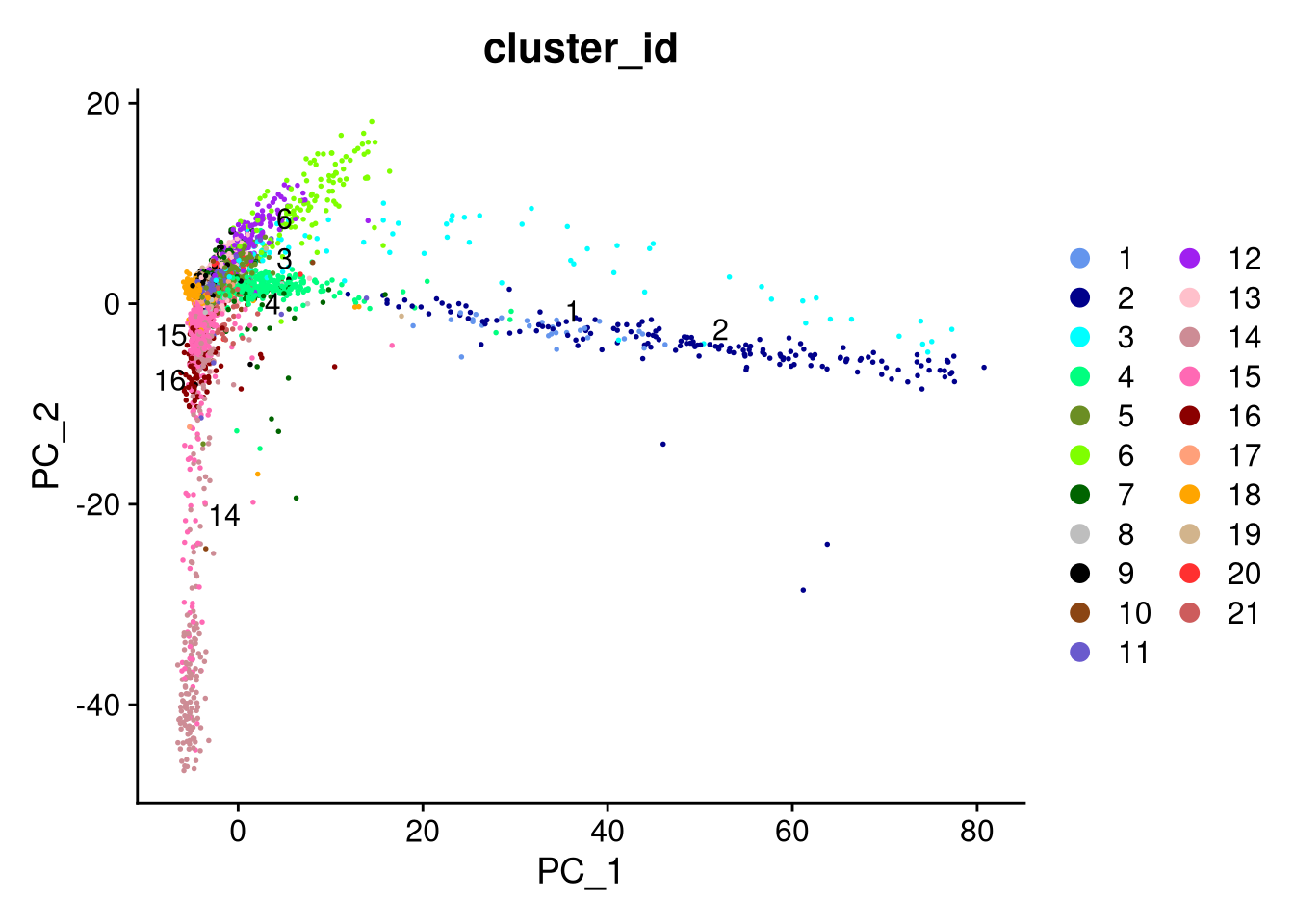
Figure 7.11: PCA plot by cluster
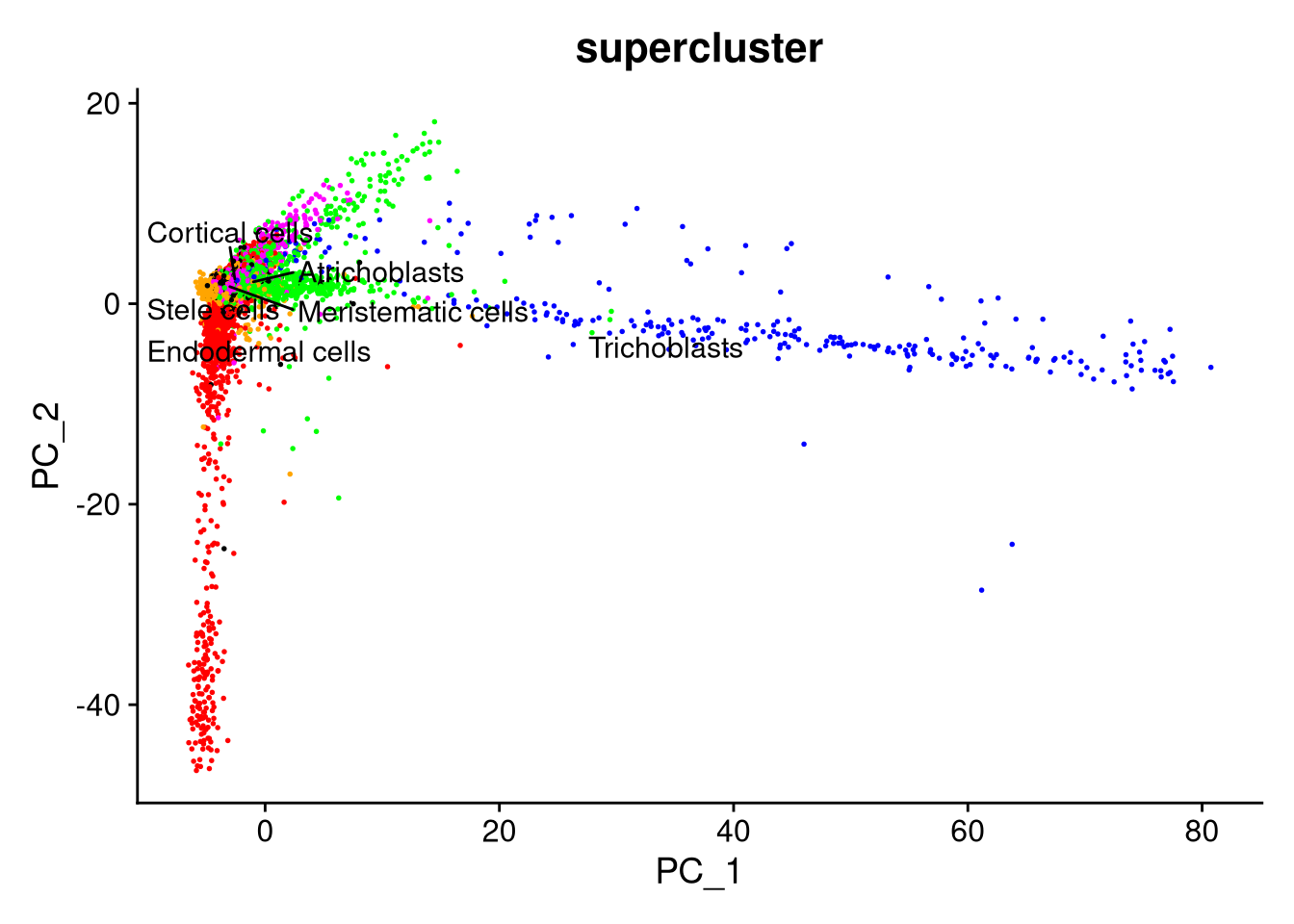
Figure 7.12: PCA plot by supercluster
We do not really need the reduction = "pca" argument in this initial example, as we have only run PCA so far. Seurat looks for existing reduced dimensions in order: first UMAP, then t-SNE, then PCA. (Therefore, we would do it this way if we wanted to visualize PCA later after having computed UMAP, for example.)
7.7.2 t-SNE
We repeat the same analysis for t-SNE, using 20 principal components.
set.seed(42)
my_seurat <- RunTSNE(my_seurat, dims = 1:20, verbose = FALSE)
my_seurat[["tsne"]]@cell.embeddings[1:6, ]## tSNE_1 tSNE_2
## AAACCCACACGCGCAT-1 9.063907 -23.306734
## AAACCCACAGAAGTTA-1 1.087900 -45.541232
## AAACCCACAGACAAAT-1 29.381223 1.189834
## AAACCCACATTGACAC-1 -21.090382 -33.508201
## AAACCCAGTAGCTTTG-1 -12.213035 -10.353685
## AAACCCAGTCCAGTTA-1 -14.058528 15.847048pdf("pdf/tsne-by-cluster-seurat.pdf")
DimPlot(my_seurat, cols = cluster_colors, group.by = "cluster_id", label.size = 4, repel = TRUE, label = TRUE)
dev.off()
pdf("pdf/tsne-by-supercluster-seurat.pdf")
DimPlot(my_seurat, cols = supercluster_colors, group.by = "supercluster", label.size = 4, repel = TRUE, label = TRUE) + NoLegend()
dev.off()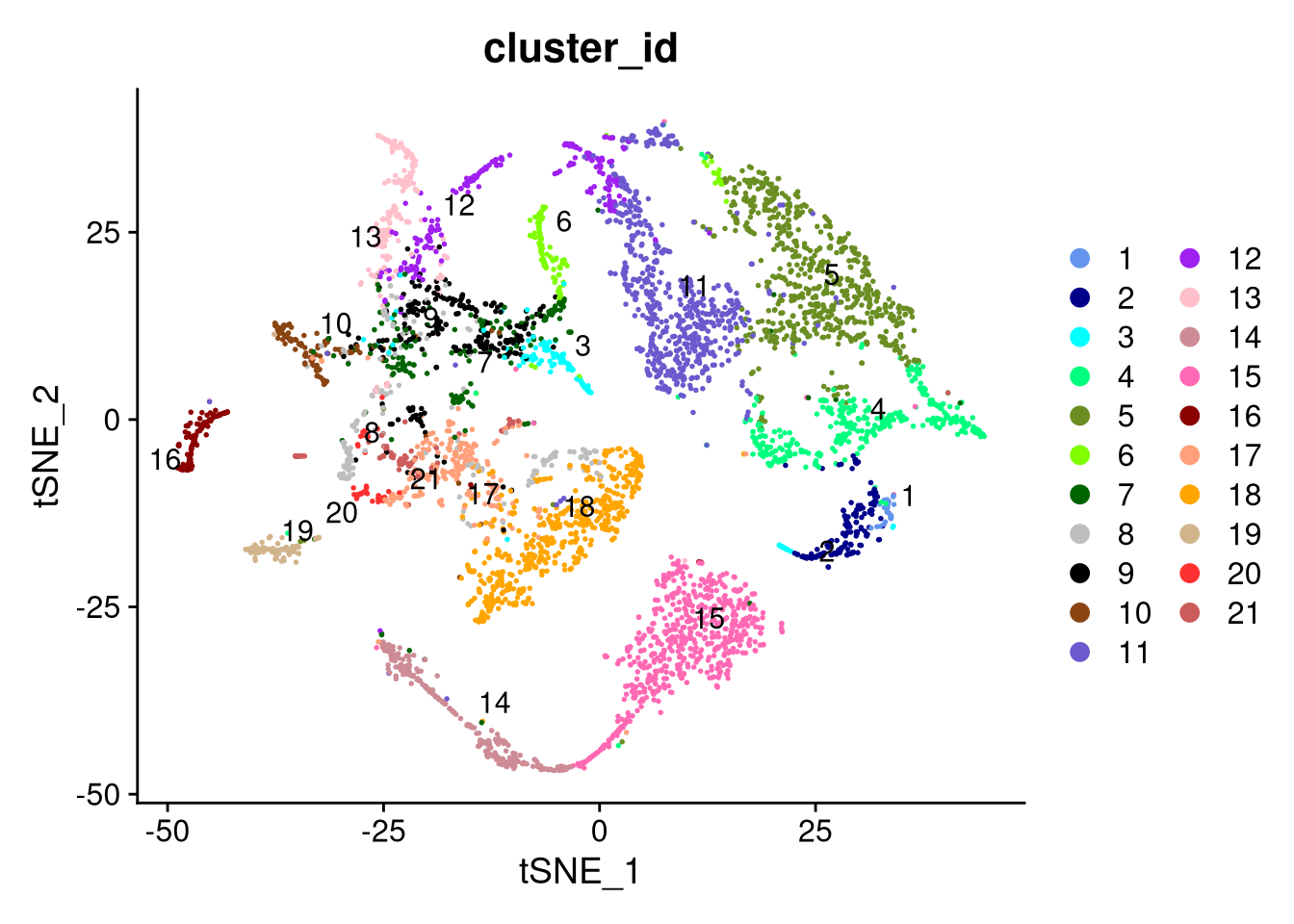
Figure 7.13: t-SNE plot by cluster
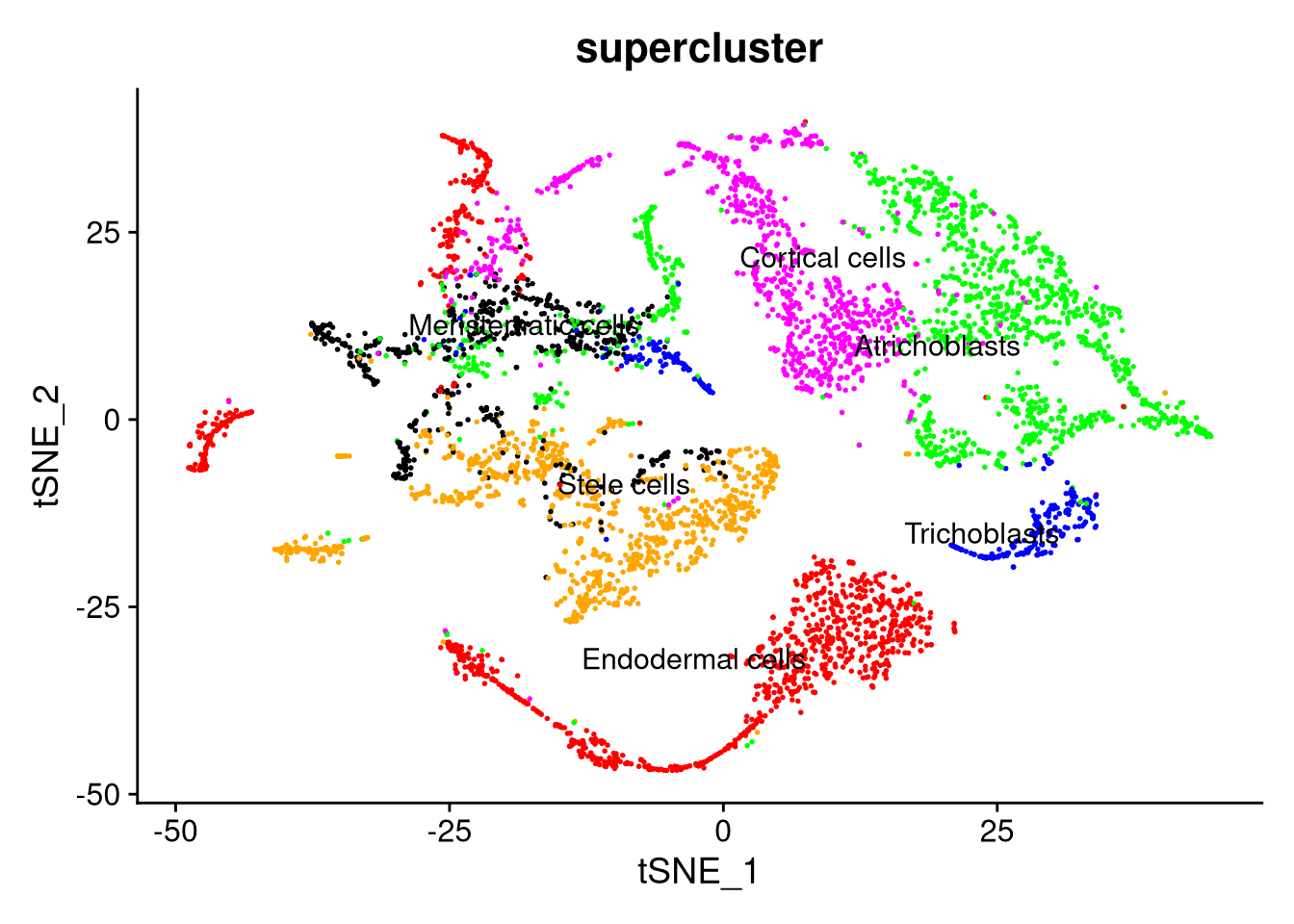
Figure 7.14: t-SNE plot by supercluster
7.7.3 UMAP
And the same for UMAP,
set.seed(42)
my_seurat <- RunUMAP(my_seurat, dims = 1:20, verbose = FALSE)
my_seurat[["umap"]]@cell.embeddings[1:6, ]## umap_1 umap_2
## AAACCCACACGCGCAT-1 -6.0059601 9.6175502
## AAACCCACAGAAGTTA-1 -10.3874970 9.5121501
## AAACCCACAGACAAAT-1 9.3276853 -0.1987486
## AAACCCACATTGACAC-1 -15.8369961 2.6514368
## AAACCCAGTAGCTTTG-1 -6.0754977 -6.1994331
## AAACCCAGTCCAGTTA-1 -0.8342648 -4.2982714pdf("pdf/umap-by-cluster-seurat.pdf")
DimPlot(my_seurat, cols = cluster_colors, group.by = "cluster_id", label.size = 4, repel = TRUE, label = TRUE)
dev.off()
pdf("pdf/umap-by-supercluster-seurat.pdf")
DimPlot(my_seurat, cols = supercluster_colors, group.by = "supercluster", label.size = 4, repel = TRUE, label = TRUE) + NoLegend()
dev.off()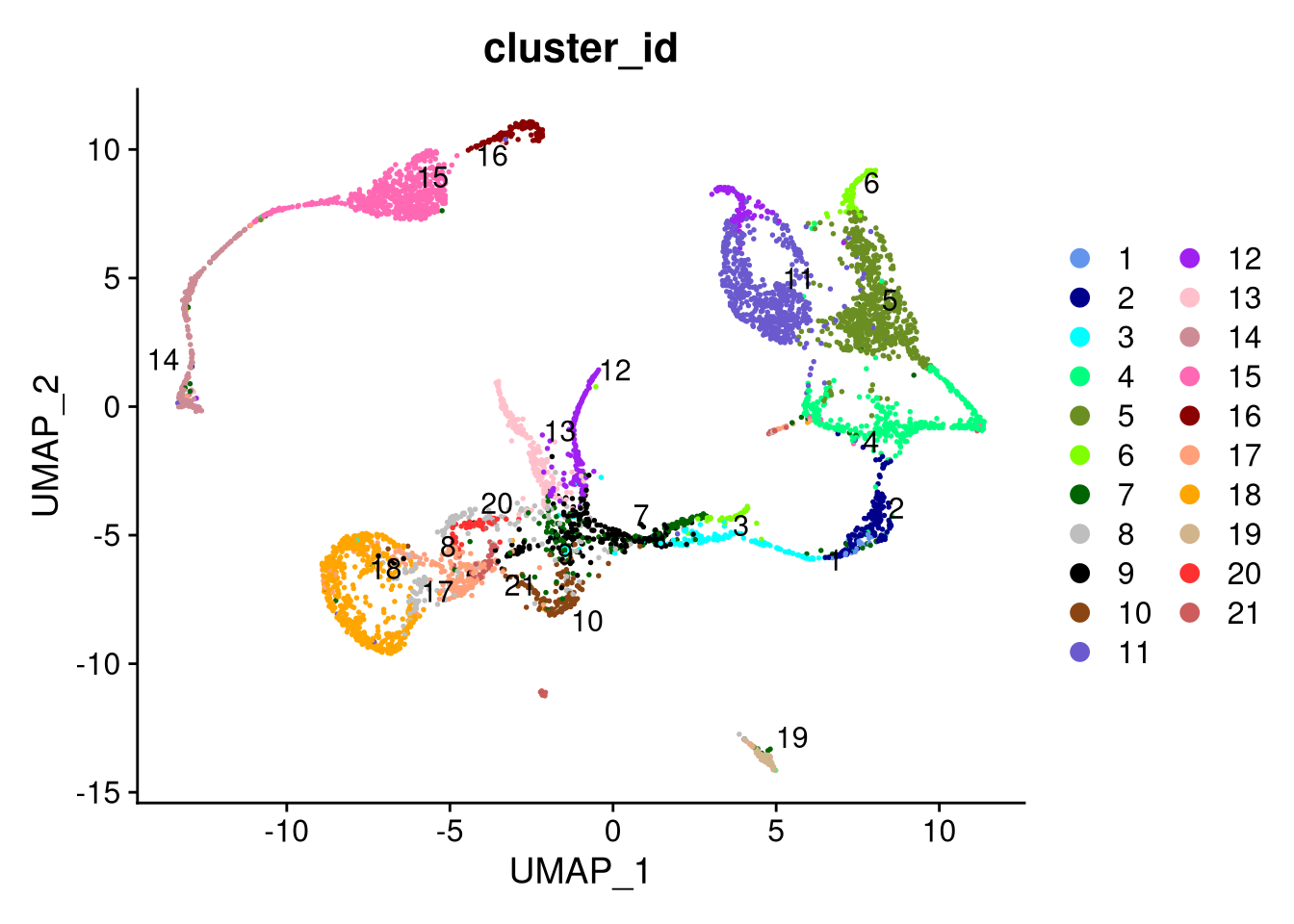
Figure 7.15: UMAP plot by cluster
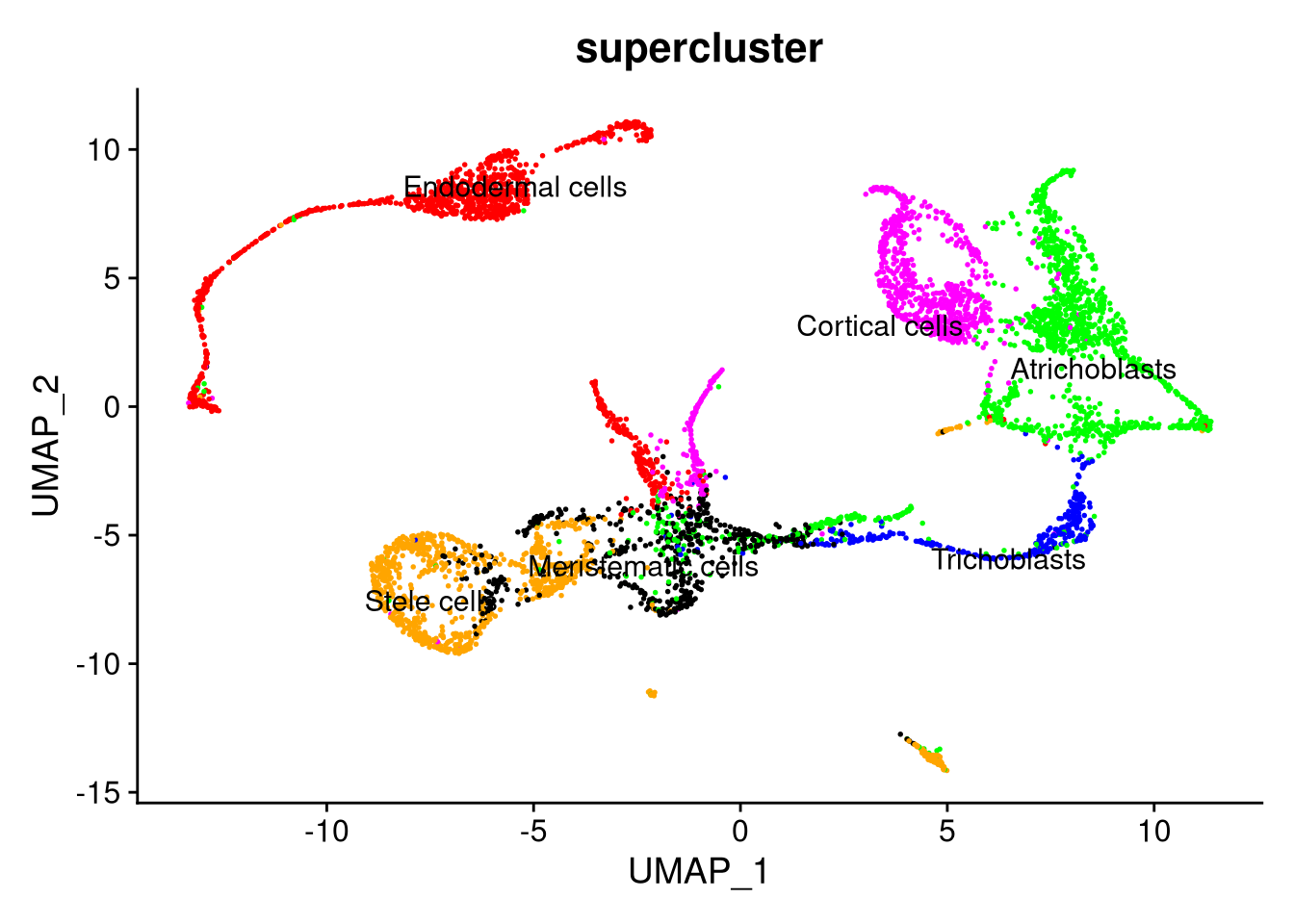
Figure 7.16: UMAP plot by supercluster
7.8 Differential gene expression
Feature plots reveal differential expression of marker genes in different cell types. Here we create feature plots for the most strongly associated gene from the first 4 principal components (from the VizDimLoadings() example above).
Idents(my_seurat) <- my_seurat$cluster_id
my_genes <- c("AT5G17390", "AT1G68850", "AT5G22310", "AT3G16340")pdf("pdf/featureplots-seurat.pdf")
FeaturePlot(my_seurat, features = my_genes, label.size = 4, repel = TRUE, label = TRUE)
dev.off()## Warning: ggrepel: 4 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## ggrepel: 4 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## ggrepel: 4 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## ggrepel: 4 unlabeled data points (too many overlaps). Consider increasing max.overlaps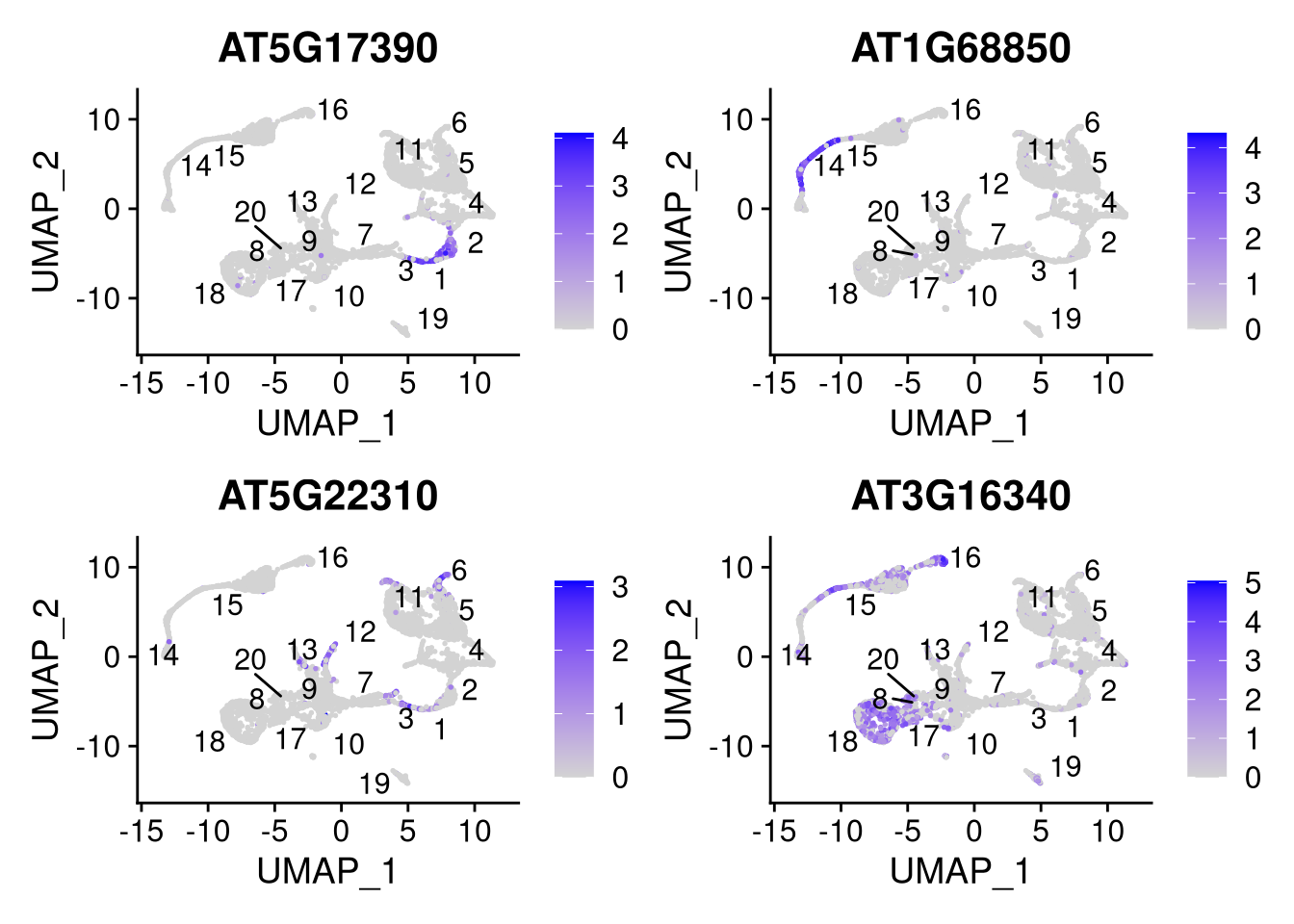
Figure 7.17: Feature plots for selected genes
An alternative is to create violin plots for genes of interest.
pdf("pdf/violin-featureplots-seurat.pdf")
VlnPlot(my_seurat, features = my_genes, cols = cluster_colors, pt.size = 0, group.by = "cluster_id", log = TRUE, ncol = 2) +
xlab("cluster_id") +
NoLegend()
dev.off()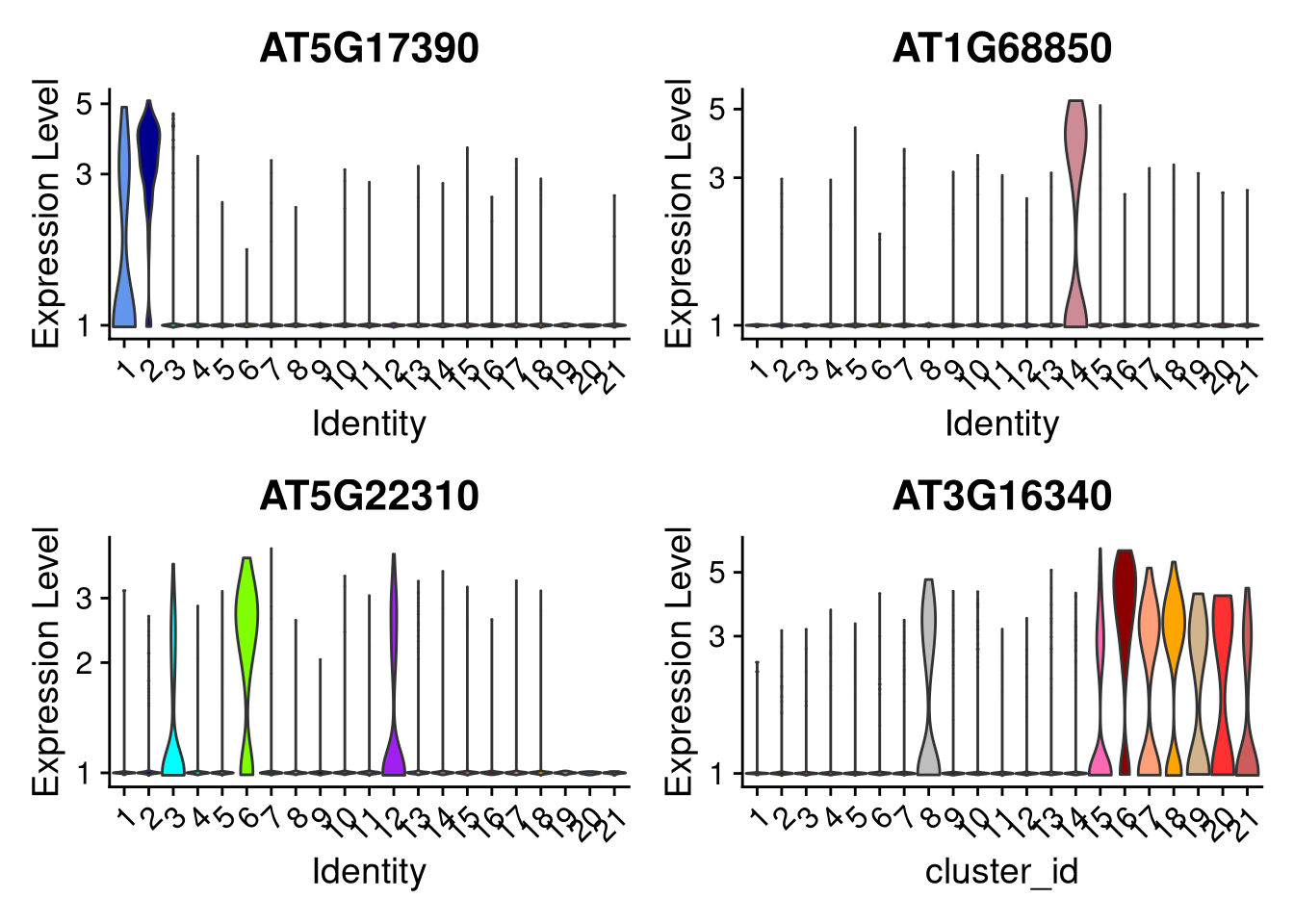
Figure 7.18: Violin plots for selected genes
Another is to make dot plots of gene expression.
pdf("pdf/dotplot-seurat.pdf")
DotPlot(my_seurat, features = top_10_genes, cols = c("darkblue", "red"), cluster.idents = TRUE, group.by = "supercluster") +
ylab("supercluster") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
dev.off()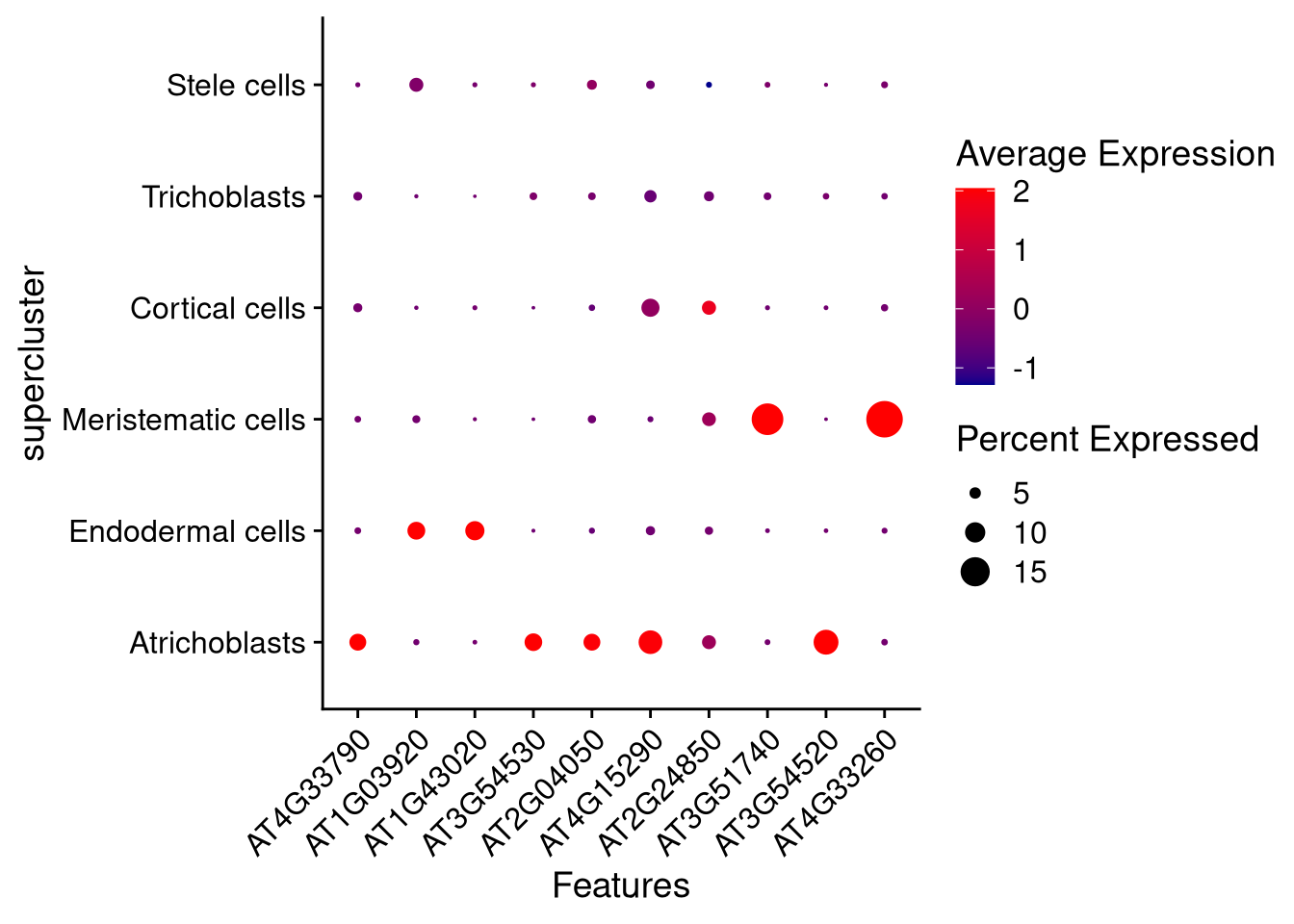
Figure 7.19: Dot plot for selected genes
Seurat also has a more direct statistical way to automatically suggest which genes go with which clusters (or any category you specify using Idents()), FindAllMarkers(). The following code identifies the top 3 genes for each cluster. First we must load the dplyr package for its data frame manipulation functions.
library(dplyr)Idents(my_seurat) <- my_seurat$cluster_id
all_markers <- FindAllMarkers(my_seurat, only.pos = TRUE, min.pct = 0.5, logfc.threshold = 0.5, verbose = FALSE)
head(all_markers)## p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
## AT2G20520 5.283152e-85 5.238077 0.750 0.037 1.259926e-80 1 AT2G20520
## AT4G08400 2.328373e-81 6.980552 0.821 0.048 5.552703e-77 1 AT4G08400
## AT3G49960 1.064398e-80 5.411229 0.929 0.063 2.538376e-76 1 AT3G49960
## AT5G04960 4.239402e-79 5.351749 0.964 0.071 1.011013e-74 1 AT5G04960
## AT5G06630 1.586581e-78 7.348940 0.929 0.067 3.783679e-74 1 AT5G06630
## AT1G12040 2.055730e-78 5.941166 0.893 0.060 4.902504e-74 1 AT1G12040top_3_markers <- as.data.frame(top_n(group_by(all_markers, cluster), n = 3, wt = avg_log2FC))
top_3_markers## p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
## 1 1.586581e-78 7.348940 0.929 0.067 3.783679e-74 1 AT5G06630
## 2 4.395165e-78 7.360210 0.786 0.045 1.048159e-73 1 AT5G35190
## 3 2.466656e-77 7.229677 0.821 0.051 5.882481e-73 1 AT4G08410
## 4 0.000000e+00 7.018590 0.802 0.006 0.000000e+00 2 AT1G70450
## 5 0.000000e+00 6.953674 0.630 0.008 0.000000e+00 2 AT4G20450
## 6 0.000000e+00 6.830985 0.525 0.003 0.000000e+00 2 AT1G08183
## 7 0.000000e+00 7.257405 0.655 0.009 0.000000e+00 3 AT1G27140
## 8 4.926469e-246 5.210145 0.543 0.017 1.174864e-241 3 AT1G66470
## 9 1.496926e-182 4.804667 0.724 0.055 3.569870e-178 3 AT2G04170
## 10 3.081863e-310 5.101303 0.616 0.050 7.349627e-306 4 AT4G28530
## 11 2.041199e-183 4.726505 0.605 0.103 4.867850e-179 4 AT4G18550
## 12 1.004522e-135 4.692208 0.562 0.118 2.395585e-131 4 AT3G48340
## 13 0.000000e+00 3.213436 0.926 0.290 0.000000e+00 5 AT1G66800
## 14 1.111775e-269 3.091744 0.842 0.264 2.651361e-265 5 AT2G37130
## 15 2.905560e-218 3.076087 0.577 0.106 6.929178e-214 5 AT3G49120
## 16 0.000000e+00 7.768793 0.633 0.008 0.000000e+00 6 AT1G06120
## 17 1.027124e-290 7.233738 0.533 0.013 2.449485e-286 6 AT2G39040
## 18 3.200221e-230 7.841542 0.600 0.026 7.631887e-226 6 AT4G33790
## 19 7.799618e-93 3.124764 0.620 0.121 1.860053e-88 7 AT1G28290
## 20 2.071338e-51 2.246058 0.572 0.168 4.939726e-47 7 AT3G62870
## 21 1.228976e-15 2.074252 0.599 0.408 2.930861e-11 7 AT1G66280
## 22 8.331900e-115 3.661119 0.541 0.072 1.986992e-110 8 AT5G44530
## 23 2.777944e-55 2.824869 0.519 0.132 6.624841e-51 8 AT1G62480
## 24 3.957489e-36 2.380275 0.546 0.190 9.437819e-32 8 AT2G16586
## 25 5.450475e-218 4.018597 0.595 0.052 1.299829e-213 9 AT3G23830
## 26 6.391708e-175 4.190787 0.547 0.058 1.524295e-170 9 AT1G09200
## 27 9.566521e-165 4.084986 0.502 0.051 2.281424e-160 9 AT1G54690
## 28 0.000000e+00 7.591374 0.703 0.009 0.000000e+00 10 AT5G01910
## 29 0.000000e+00 8.147332 0.602 0.005 0.000000e+00 10 AT5G55520
## 30 1.244835e-232 7.698745 0.542 0.020 2.968683e-228 10 AT4G33270
## 31 0.000000e+00 4.860840 0.559 0.027 0.000000e+00 11 AT2G29320
## 32 1.035624e-297 3.942144 0.611 0.075 2.469757e-293 11 AT1G65970
## 33 1.435710e-297 4.089394 0.561 0.057 3.423882e-293 11 AT4G15393
## 34 8.658822e-184 4.797775 0.588 0.061 2.064956e-179 12 AT5G49360
## 35 8.875062e-181 3.599919 0.575 0.055 2.116525e-176 12 AT2G02080
## 36 1.947868e-109 3.656949 0.570 0.104 4.645277e-105 12 AT3G23470
## 37 0.000000e+00 6.086744 0.695 0.026 0.000000e+00 13 AT2G14900
## 38 0.000000e+00 7.112965 0.658 0.022 0.000000e+00 13 AT4G11290
## 39 0.000000e+00 6.424532 0.500 0.011 0.000000e+00 13 AT5G45200
## 40 0.000000e+00 8.645667 0.681 0.006 0.000000e+00 14 AT2G24430
## 41 0.000000e+00 7.902543 0.500 0.010 0.000000e+00 14 AT3G48450
## 42 3.694241e-209 7.937899 0.532 0.045 8.810027e-205 14 AT5G37690
## 43 0.000000e+00 5.576704 0.859 0.056 0.000000e+00 15 AT2G28470
## 44 0.000000e+00 5.064288 0.753 0.039 0.000000e+00 15 AT1G14160
## 45 0.000000e+00 5.830053 0.637 0.035 0.000000e+00 15 AT2G29330
## 46 0.000000e+00 9.429126 0.766 0.005 0.000000e+00 16 AT5G51680
## 47 0.000000e+00 9.060124 0.757 0.015 0.000000e+00 16 AT1G43020
## 48 0.000000e+00 8.843023 0.505 0.007 0.000000e+00 16 AT1G44970
## 49 3.959335e-197 3.913483 0.751 0.102 9.442222e-193 17 AT1G75500
## 50 1.905619e-146 3.818736 0.631 0.096 4.544521e-142 17 AT2G02100
## 51 6.921911e-141 4.247002 0.524 0.065 1.650737e-136 17 AT3G53100
## 52 0.000000e+00 5.629972 0.831 0.044 0.000000e+00 18 AT1G31050
## 53 0.000000e+00 5.989980 0.779 0.038 0.000000e+00 18 AT3G62040
## 54 3.359451e-282 5.924693 0.563 0.063 8.011618e-278 18 AT1G32450
## 55 0.000000e+00 9.259887 0.919 0.013 0.000000e+00 19 AT5G64240
## 56 0.000000e+00 9.304102 0.837 0.017 0.000000e+00 19 AT4G00670
## 57 0.000000e+00 9.624342 0.558 0.003 0.000000e+00 19 AT4G01600
## 58 1.239069e-278 7.667238 0.667 0.009 2.954932e-274 20 AT5G03960
## 59 3.463598e-177 7.121955 0.548 0.011 8.259989e-173 20 AT3G59370
## 60 4.528058e-128 6.328608 0.738 0.033 1.079851e-123 20 AT1G12080
## 61 0.000000e+00 7.296757 0.512 0.004 0.000000e+00 21 AT5G66300
## 62 3.591989e-214 6.080467 0.610 0.021 8.566176e-210 21 AT1G47410
## 63 1.691360e-116 5.286322 0.512 0.031 4.033555e-112 21 AT5G19530Click for answer
Idents(my_seurat) <- my_seurat$supercluster
all_markers <- FindAllMarkers(my_seurat, only.pos = TRUE, min.pct = 0.5, logfc.threshold = 0.5, verbose = FALSE)
top_10_markers <- as.data.frame(top_n(group_by(all_markers, cluster), n = 10, wt = avg_log2FC))
top_10_markers## p_val avg_log2FC pct.1 pct.2 p_val_adj cluster
## 1 0.000000e+00 7.288049 0.683 0.045 0.000000e+00 Trichoblasts
## 2 0.000000e+00 6.897367 0.654 0.021 0.000000e+00 Trichoblasts
## 3 0.000000e+00 7.561357 0.611 0.012 0.000000e+00 Trichoblasts
## 4 0.000000e+00 6.741956 0.598 0.019 0.000000e+00 Trichoblasts
## 5 0.000000e+00 6.850448 0.592 0.013 0.000000e+00 Trichoblasts
## 6 0.000000e+00 7.249377 0.556 0.008 0.000000e+00 Trichoblasts
## 7 0.000000e+00 7.198315 0.536 0.006 0.000000e+00 Trichoblasts
## 8 0.000000e+00 7.659366 0.533 0.004 0.000000e+00 Trichoblasts
## 9 0.000000e+00 7.112747 0.516 0.005 0.000000e+00 Trichoblasts
## 10 0.000000e+00 6.686506 0.513 0.007 0.000000e+00 Trichoblasts
## 11 0.000000e+00 3.471082 0.673 0.124 0.000000e+00 Atrichoblasts
## 12 0.000000e+00 2.918668 0.704 0.180 0.000000e+00 Atrichoblasts
## 13 2.057269e-315 3.467894 0.614 0.124 4.906175e-311 Atrichoblasts
## 14 1.072408e-273 2.972052 0.577 0.115 2.557477e-269 Atrichoblasts
## 15 2.933857e-270 3.035185 0.704 0.255 6.996661e-266 Atrichoblasts
## 16 8.836606e-268 3.257136 0.676 0.219 2.107354e-263 Atrichoblasts
## 17 1.492579e-253 3.046109 0.510 0.087 3.559503e-249 Atrichoblasts
## 18 6.477013e-249 2.902410 0.632 0.190 1.544638e-244 Atrichoblasts
## 19 1.674176e-240 2.898963 0.680 0.229 3.992575e-236 Atrichoblasts
## 20 1.716924e-239 3.100709 0.529 0.106 4.094520e-235 Atrichoblasts
## 21 1.428019e-211 3.416145 0.609 0.121 3.405540e-207 Meristematic cells
## 22 4.040242e-201 2.908276 0.725 0.208 9.635169e-197 Meristematic cells
## 23 4.390477e-178 2.739719 0.711 0.221 1.047041e-173 Meristematic cells
## 24 2.750088e-172 3.065536 0.745 0.290 6.558409e-168 Meristematic cells
## 25 3.928823e-162 2.797085 0.591 0.145 9.369458e-158 Meristematic cells
## 26 4.234139e-162 2.759429 0.616 0.161 1.009757e-157 Meristematic cells
## 27 4.341591e-158 2.723544 0.600 0.155 1.035383e-153 Meristematic cells
## 28 1.295380e-146 2.725525 0.533 0.122 3.089222e-142 Meristematic cells
## 29 3.287890e-138 2.761369 0.513 0.122 7.840960e-134 Meristematic cells
## 30 1.278088e-131 2.785672 0.591 0.187 3.047985e-127 Meristematic cells
## 31 0.000000e+00 4.302087 0.592 0.036 0.000000e+00 Cortical cells
## 32 1.338972e-314 2.724336 0.803 0.200 3.193180e-310 Cortical cells
## 33 4.320275e-287 2.949479 0.578 0.077 1.030299e-282 Cortical cells
## 34 3.986864e-278 3.387030 0.542 0.071 9.507873e-274 Cortical cells
## 35 1.113431e-266 3.021412 0.558 0.082 2.655310e-262 Cortical cells
## 36 4.732985e-247 2.928614 0.581 0.103 1.128722e-242 Cortical cells
## 37 2.324000e-244 2.703855 0.639 0.138 5.542274e-240 Cortical cells
## 38 1.599894e-240 2.765176 0.817 0.301 3.815427e-236 Cortical cells
## 39 3.937027e-192 2.622235 0.668 0.213 9.389021e-188 Cortical cells
## 40 2.001277e-179 2.934241 0.569 0.150 4.772645e-175 Cortical cells
## 41 0.000000e+00 5.311556 0.911 0.213 0.000000e+00 Endodermal cells
## 42 0.000000e+00 5.683594 0.633 0.058 0.000000e+00 Endodermal cells
## 43 0.000000e+00 5.273478 0.645 0.080 0.000000e+00 Endodermal cells
## 44 0.000000e+00 5.572345 0.554 0.041 0.000000e+00 Endodermal cells
## 45 0.000000e+00 4.864676 0.588 0.086 0.000000e+00 Endodermal cells
## 46 0.000000e+00 4.707194 0.544 0.043 0.000000e+00 Endodermal cells
## 47 0.000000e+00 5.343380 0.582 0.085 0.000000e+00 Endodermal cells
## 48 0.000000e+00 5.956437 0.525 0.044 0.000000e+00 Endodermal cells
## 49 0.000000e+00 6.106374 0.512 0.032 0.000000e+00 Endodermal cells
## 50 0.000000e+00 5.188181 0.512 0.046 0.000000e+00 Endodermal cells
## 51 0.000000e+00 4.534503 0.804 0.082 0.000000e+00 Stele cells
## 52 0.000000e+00 4.877146 0.569 0.053 0.000000e+00 Stele cells
## 53 0.000000e+00 5.188296 0.507 0.037 0.000000e+00 Stele cells
## 54 5.860327e-291 3.664002 0.599 0.108 1.397571e-286 Stele cells
## 55 4.894555e-266 4.724278 0.501 0.075 1.167253e-261 Stele cells
## 56 1.972447e-192 2.734800 0.598 0.183 4.703893e-188 Stele cells
## 57 5.662993e-179 3.857542 0.535 0.163 1.350511e-174 Stele cells
## 58 9.108284e-150 2.309007 0.524 0.159 2.172144e-145 Stele cells
## 59 3.720749e-147 2.859875 0.645 0.308 8.873243e-143 Stele cells
## 60 1.243051e-104 2.664079 0.504 0.224 2.964428e-100 Stele cells
## gene
## 1 AT1G12560
## 2 AT5G40860
## 3 AT5G17390
## 4 AT5G19560
## 5 AT3G12540
## 6 AT2G03720
## 7 AT1G79860
## 8 AT1G12150
## 9 AT2G35890
## 10 AT1G34760
## 11 AT3G12977
## 12 AT3G09220
## 13 AT1G73300
## 14 AT2G29995
## 15 AT1G66800
## 16 AT2G37130
## 17 AT5G18270
## 18 AT1G14960
## 19 AT4G15230
## 20 AT1G05320
## 21 AT1G56110
## 22 AT4G17390
## 23 AT3G60245
## 24 AT1G29418
## 25 AT2G01250
## 26 AT1G52300
## 27 AT2G27710
## 28 AT4G26230
## 29 AT2G19730
## 30 AT1G48920
## 31 AT3G46500
## 32 AT5G42250
## 33 AT5G65380
## 34 AT4G36220
## 35 AT3G26590
## 36 AT4G36870
## 37 AT3G51860
## 38 AT4G30170
## 39 AT4G30270
## 40 AT4G02520
## 41 AT3G32980
## 42 AT1G05260
## 43 AT3G22600
## 44 AT5G41040
## 45 AT1G61590
## 46 AT5G23920
## 47 AT1G04220
## 48 AT2G48130
## 49 AT1G08670
## 50 AT2G28470
## 51 AT4G34600
## 52 AT2G40900
## 53 AT1G31050
## 54 AT2G43910
## 55 AT4G01450
## 56 AT3G14990
## 57 AT5G23050
## 58 AT4G24060
## 59 AT5G59780
## 60 AT1G69850matching_markers <- intersect(top_3_markers$gene, top_10_markers$gene)
matching_markers## [1] "AT1G66800" "AT2G37130" "AT2G28470" "AT1G31050"7.9 Clustering
We can use SingleR to predict cell types for a Seurat object, as we did for a SingleCellExperiment. However, instead of passing a Seurat object as the test or ref argument, we must pass its normalized counts matrix (data).
library(SingleR)
another_seurat <- readRDS("data/another_seurat.rds")
predicted_clusters <- SingleR(test = another_seurat[["RNA"]]$data, ref = my_seurat[["RNA"]]$data, labels = my_seurat$cluster_id)Seurat can also assign clusters for you. Its FindClusters() function does not use k-means as in the previous chapter, but a shared nearest neighbor clustering algorithm. We need not specify a number of desired clusters.
my_seurat <- FindNeighbors(my_seurat, dims = 1:10, verbose = FALSE)
my_seurat <- FindClusters(my_seurat, resolution = 0.5, verbose = FALSE)
table(my_seurat$seurat_clusters)##
## 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
## 650 540 521 511 483 467 418 347 248 215 192 134 132 116 91 86pdf("pdf/umap-by-computed-cluster-seurat.pdf")
DimPlot(my_seurat, label.size = 4, repel = TRUE, label = TRUE)
dev.off()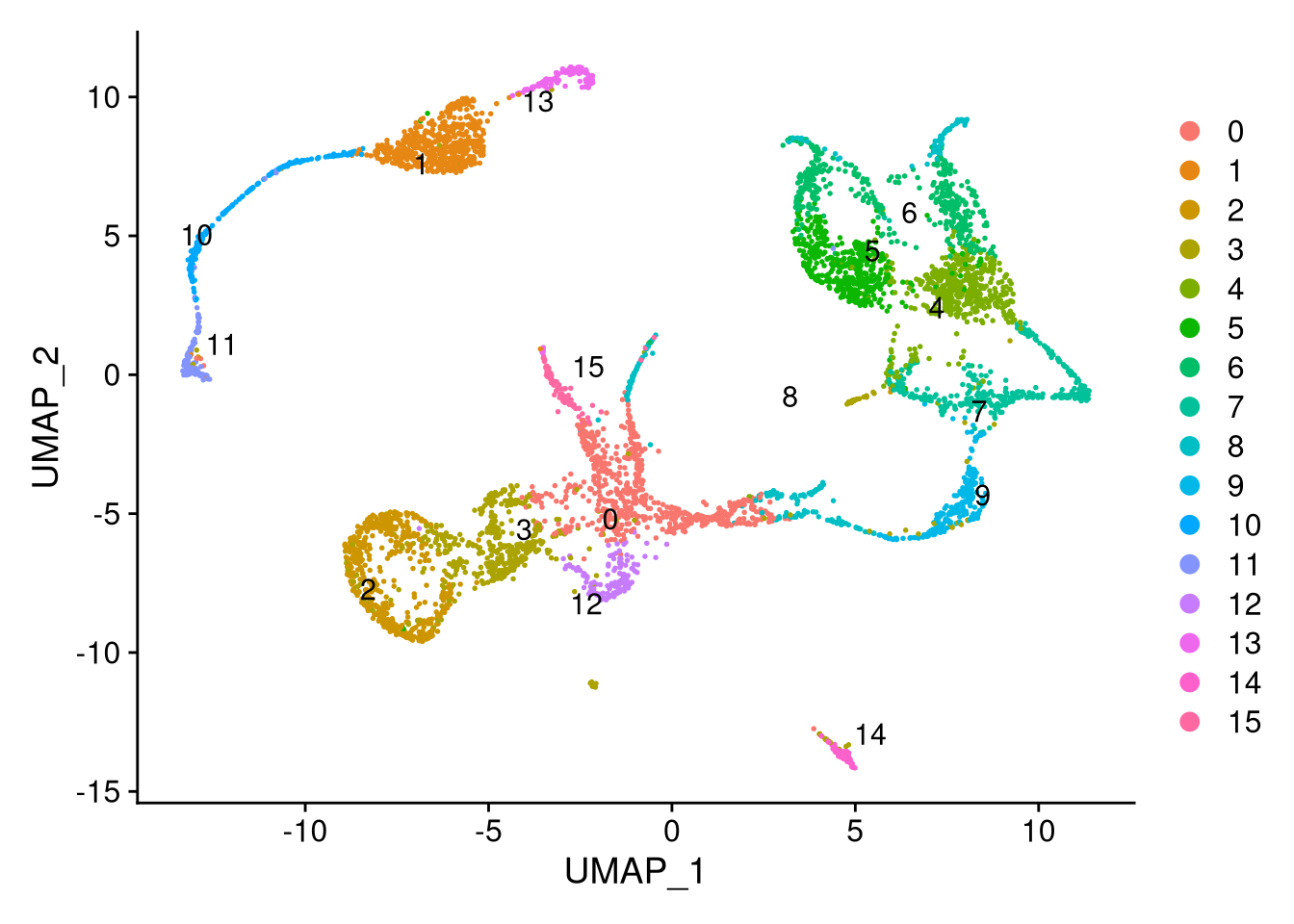
Figure 7.20: UMAP plot by computed cluster
This method groups our cells into 16 clusters instead of 21. Note that they start from 0 and go in the seurat_clusters column of the metadata.
7.10 SCTransform normalization
The SCTransform() function (in package Seurat) provides an alternative to log-normalization, based on regularized negative binomial regression. It is designed to better capture the variation due to biology over spurious variation.
my_seurat <- SCTransform(my_seurat, ncells = ncol(my_seurat), verbose = FALSE)
my_seurat## An object of class Seurat
## 45005 features across 5151 samples within 2 assays
## Active assay: SCT (21157 features, 3000 variable features)
## 3 layers present: counts, data, scale.data
## 1 other assay present: RNA
## 3 dimensional reductions calculated: pca, tsne, umapNote that SCTransform() replaces all three of NormalizeData(), ScaleData(), and FindVariableFeatures(); you do not need to run the latter two separately.
We have created a new assay called “SCT” and made it the active (default) one. Now we can recompute the reduced dimensions based on SCTransform.
my_seurat <- RunPCA(my_seurat, verbose = FALSE)
my_seurat <- RunUMAP(my_seurat, dims = 1:20, verbose = FALSE)pdf("pdf/umap-by-cluster-sct-seurat.pdf")
DimPlot(my_seurat, cols = cluster_colors, group.by = "cluster_id", label.size = 4, repel = TRUE, label = TRUE)
dev.off()
pdf("pdf/umap-by-supercluster-sct-seurat.pdf")
DimPlot(my_seurat, cols = supercluster_colors, group.by = "supercluster", label.size = 4, repel = TRUE, label = TRUE) + NoLegend()
dev.off()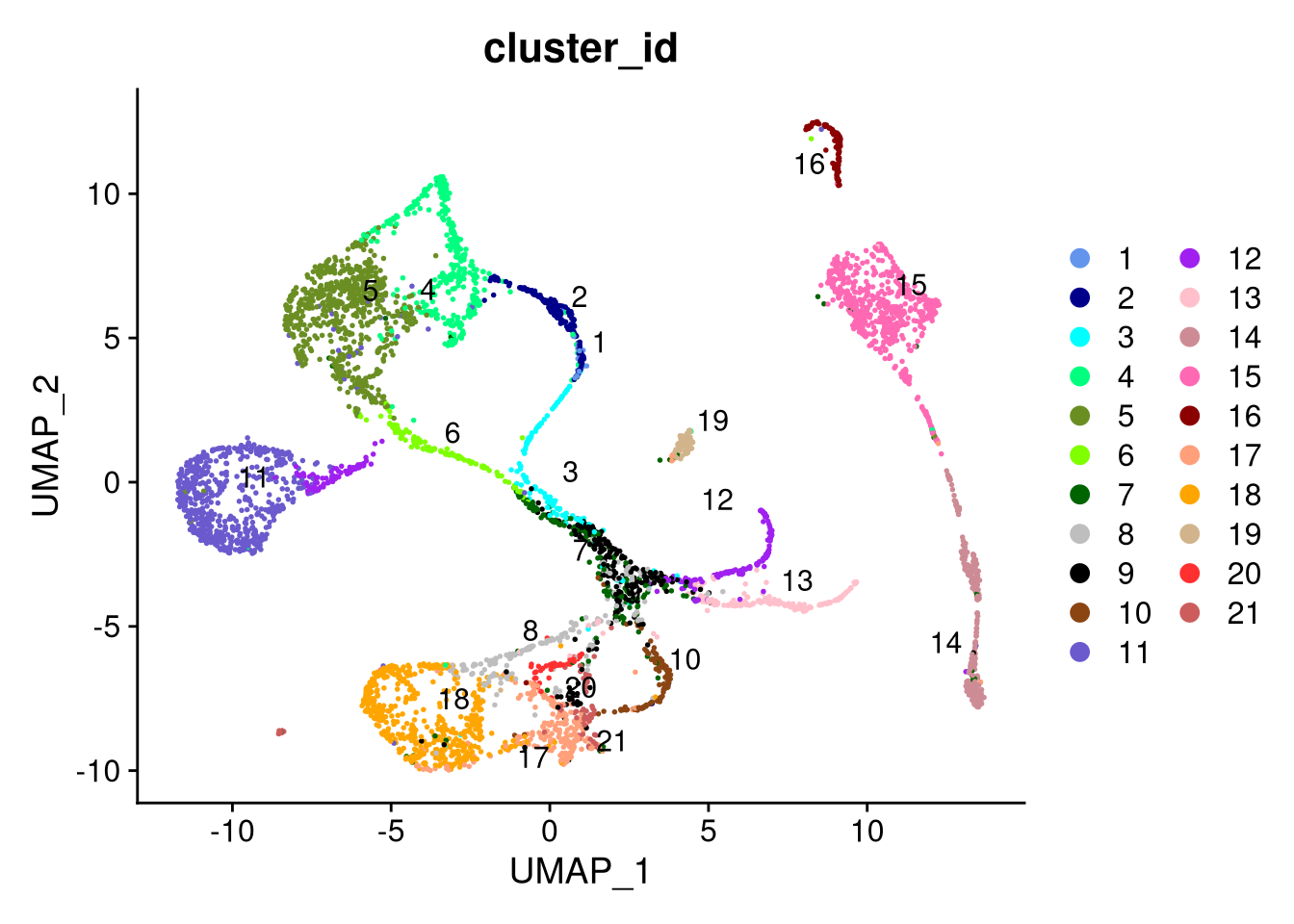
Figure 7.21: UMAP plot by cluster, using SCTransform normalization
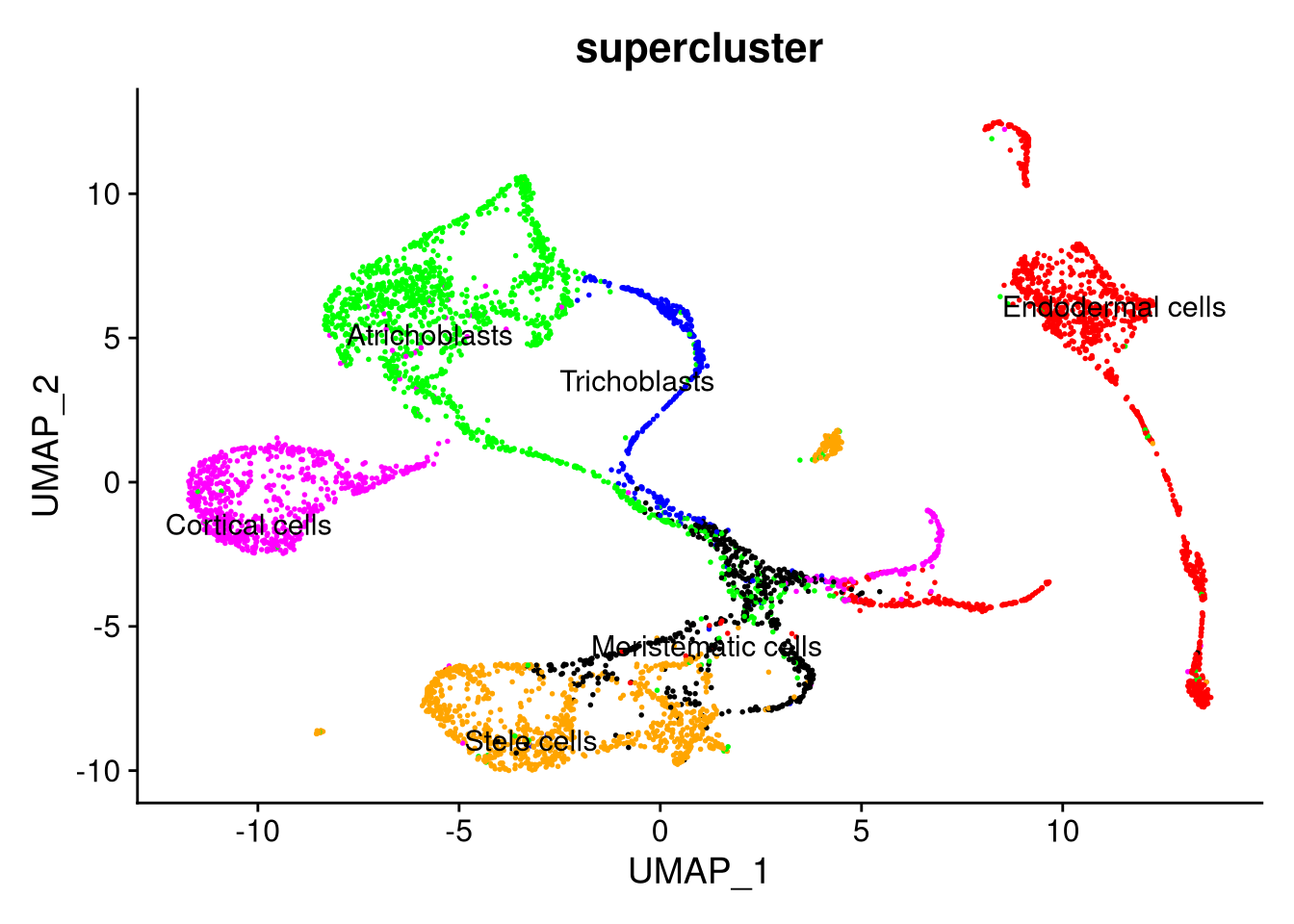
Figure 7.22: UMAP plot by supercluster, using SCTransform normalization
SCTransform hold that after normalizing this way, we should use more principal components when running UMAP. Try it with 40 PCs (dimensions) instead of 20. What do you think?
Click for answer
Rerun UMAP with 40 PCs, then make the dimensionality reduction charts.
my_seurat <- RunUMAP(my_seurat, dims = 1:40, verbose = FALSE)pdf("pdf/umap-by-cluster-sct-seurat-40-pcs.pdf")
DimPlot(my_seurat, cols = cluster_colors, group.by = "cluster_id", label.size = 4, repel = TRUE, label = TRUE)
dev.off()
pdf("pdf/umap-by-supercluster-sct-seurat-40-pcs.pdf")
DimPlot(my_seurat, cols = supercluster_colors, group.by = "supercluster", label.size = 4, repel = TRUE, label = TRUE) + NoLegend()
dev.off()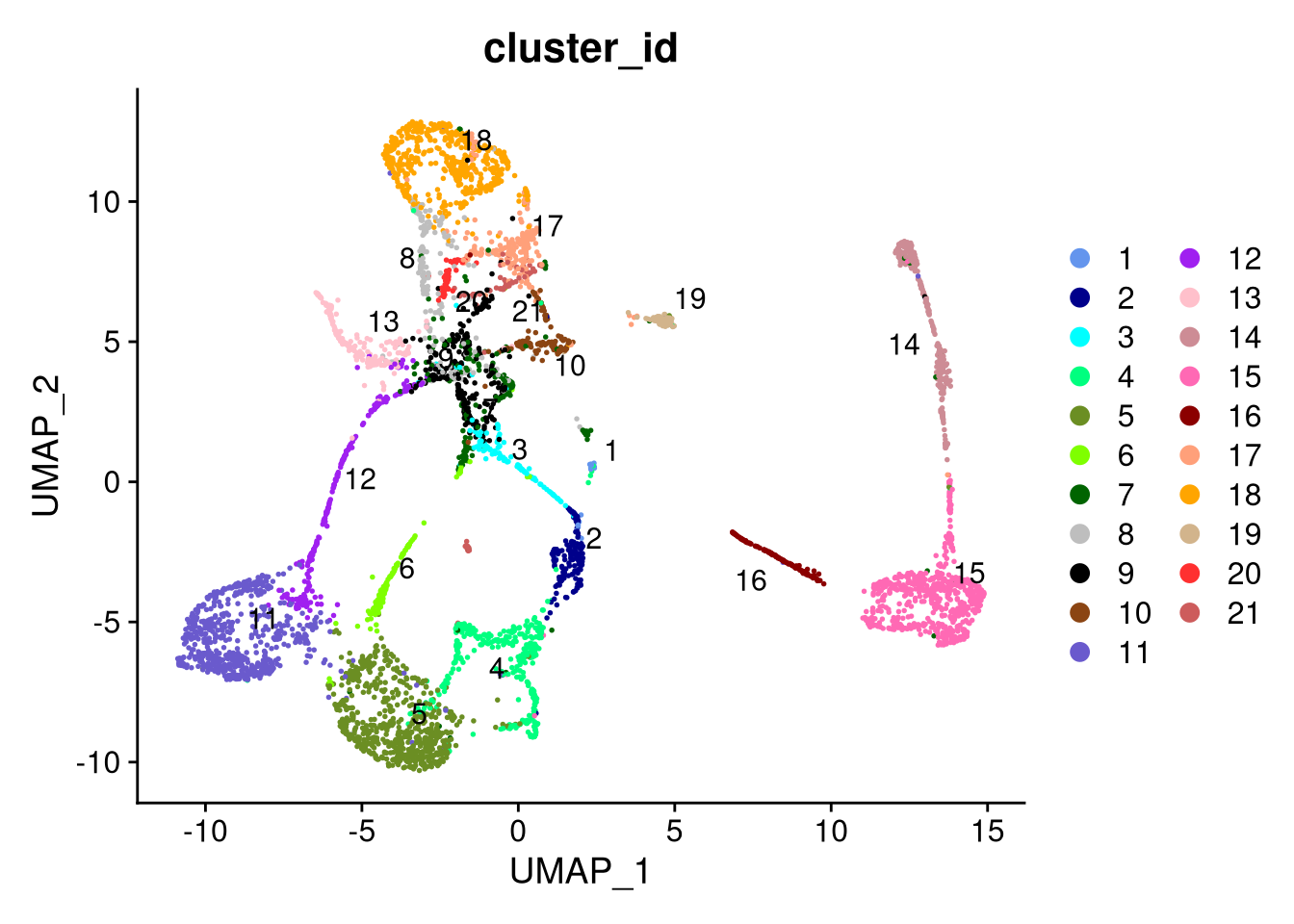
Figure 7.23: UMAP plot by cluster, using SCTransform normalization with 40 PCs
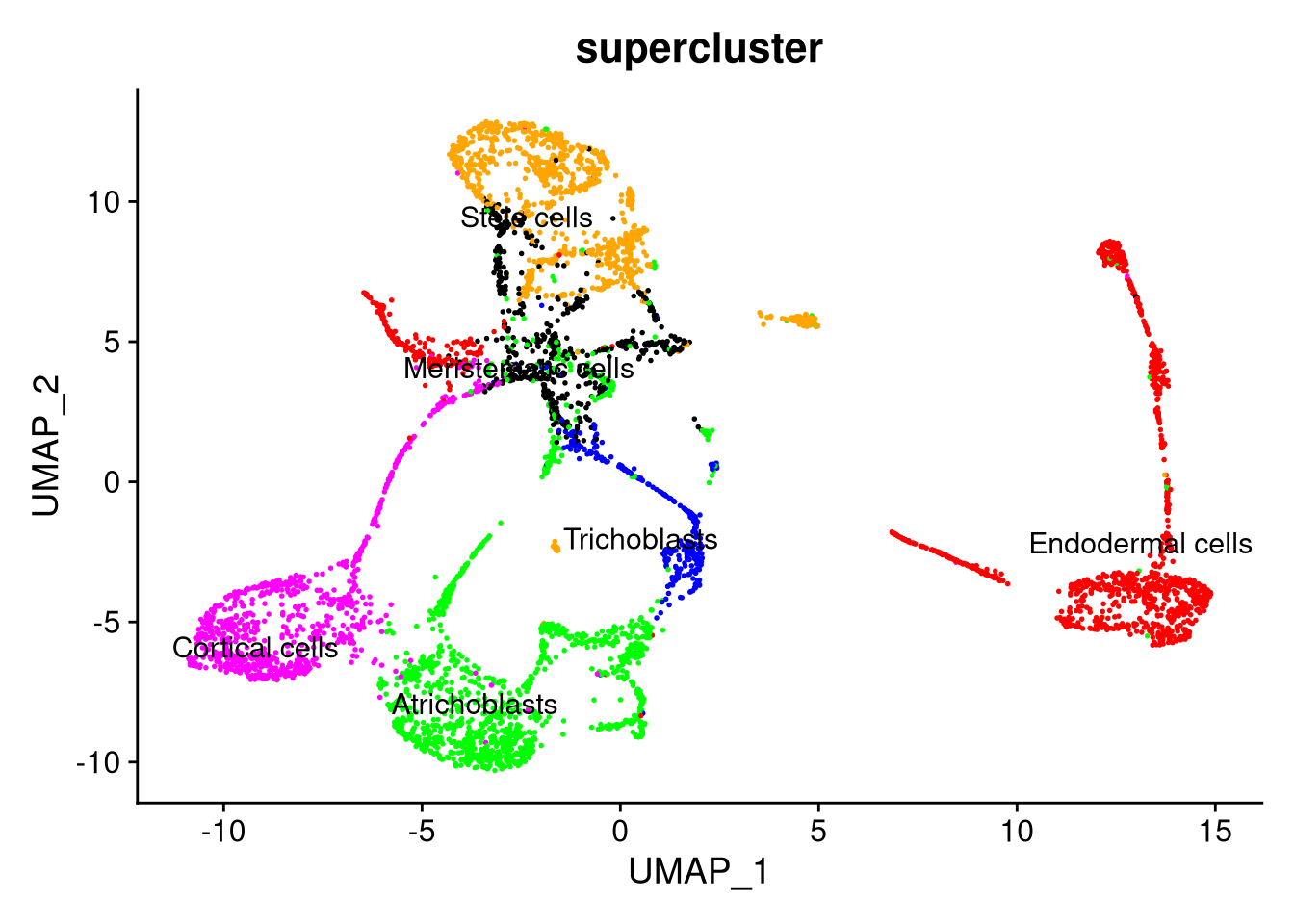
Figure 7.24: UMAP plot by supercluster, using SCTransform normalization with 40 PCs
Note that the PCA and UMAP dimensionality reductions now live in the “SCT” assay instead of the original “RNA” assay, leaving only t-SNE in “RNA”. If you wanted to recreate our initial PCA or UMAP plots, you would have to change the active assay back to “RNA” and recompute them there (or reload my_seurat from a previously saved state).
my_seurat@reductions## $pca
## A dimensional reduction object with key PC_
## Number of dimensions: 50
## Number of cells: 5151
## Projected dimensional reduction calculated: FALSE
## Jackstraw run: FALSE
## Computed using assay: SCT
##
## $tsne
## A dimensional reduction object with key tSNE_
## Number of dimensions: 2
## Number of cells: 5151
## Projected dimensional reduction calculated: FALSE
## Jackstraw run: FALSE
## Computed using assay: RNA
##
## $umap
## A dimensional reduction object with key umap_
## Number of dimensions: 2
## Number of cells: 5151
## Projected dimensional reduction calculated: FALSE
## Jackstraw run: FALSE
## Computed using assay: SCT7.11 Conversion between Seurat and SingleCellExperiment
After trying both SingleCellExperiment and Seurat, you may develop a preference for one of them and end up using it in your work. In case a collaborator shares a dataset in the other format, you need to be able to convert it to yours. You may also want to use functionality that exists in only one format.
The as.SingleCellExperiment() function (from package Seurat) provides a quick way to convert an existing Seurat object to SingleCellExperiment. (We will of course need to reload the SingleCellExperiment package.)
library(SingleCellExperiment)fake_SCE_2 <- as.SingleCellExperiment(fake_seurat)
counts(fake_SCE_2)## 12 x 8 sparse Matrix of class "dgCMatrix"
## Cell-1 Cell-2 Cell-3 Cell-4 Cell-5 Cell-6 Cell-7 Cell-8
## Gene-1 . . 2 . . . 1 2
## Gene-2 1 . 1 . 13 2 1 .
## Gene-3 . 2 2 . . . 2 .
## Gene-4 . . . 3 3 . 13 .
## Gene-5 . . . 1 2 . 2 .
## Gene-6 2 1 2 1 . . . 1
## Gene-7 . 2 1 . 1 3 1 5
## Gene-8 . . 6 2 . 1 2 3
## Gene-9 2 9 . 3 2 . 2 4
## Gene-10 1 1 . . 1 . . 2
## Gene-11 2 9 3 8 . 11 . 1
## Gene-12 4 . 1 2 16 . . 3logcounts(fake_SCE_2)## 12 x 8 sparse Matrix of class "dgCMatrix"
## Cell-1 Cell-2 Cell-3 Cell-4 Cell-5 Cell-6 Cell-7 Cell-8
## Gene-1 . . 7.014015 . . . 6.034684 6.860015
## Gene-2 6.726633 . 6.321767 . 8.137996 7.071124 6.034684 .
## Gene-3 . 6.726633 7.014015 . . . 6.726633 .
## Gene-4 . . . 7.313887 6.672632 . 8.597420 .
## Gene-5 . . . 6.216606 6.267800 . 6.726633 .
## Gene-6 7.419181 6.034684 7.014015 6.216606 . . . 6.167916
## Gene-7 . 6.726633 6.321767 . 5.576547 7.476306 6.034684 7.775676
## Gene-8 . . 8.112028 6.908755 . 6.378826 6.726633 7.265130
## Gene-9 7.419181 8.229778 . 7.313887 6.267800 . 6.726633 7.552637
## Gene-10 6.726633 6.034684 . . 5.576547 . . 6.860015
## Gene-11 7.419181 8.229778 7.419181 8.294300 . 8.775177 . 6.167916
## Gene-12 8.112028 . 6.321767 6.908755 8.345580 . . 7.265130colData(fake_SCE_2)## DataFrame with 8 rows and 6 columns
## orig.ident nCount_RNA nFeature_RNA nCount_CPM nFeature_CPM ident
## <factor> <numeric> <integer> <numeric> <integer> <factor>
## Cell-1 Fake Seurat 12 6 12 6 Fake Seurat
## Cell-2 Fake Seurat 24 6 24 6 Fake Seurat
## Cell-3 Fake Seurat 18 8 18 8 Fake Seurat
## Cell-4 Fake Seurat 20 7 20 7 Fake Seurat
## Cell-5 Fake Seurat 38 7 38 7 Fake Seurat
## Cell-6 Fake Seurat 17 4 17 4 Fake Seurat
## Cell-7 Fake Seurat 24 8 24 8 Fake Seurat
## Cell-8 Fake Seurat 21 8 21 8 Fake SeuratSimilarly, the as.Seurat() function (from package SeuratObject) converts a SingleCellExperiment object to Seurat.
fake_seurat_2 <- as.Seurat(fake_SCE_2)
fake_seurat_2[["RNA"]]$counts## 12 x 8 sparse Matrix of class "dgCMatrix"
## Cell-1 Cell-2 Cell-3 Cell-4 Cell-5 Cell-6 Cell-7 Cell-8
## Gene-1 . . 2 . . . 1 2
## Gene-2 1 . 1 . 13 2 1 .
## Gene-3 . 2 2 . . . 2 .
## Gene-4 . . . 3 3 . 13 .
## Gene-5 . . . 1 2 . 2 .
## Gene-6 2 1 2 1 . . . 1
## Gene-7 . 2 1 . 1 3 1 5
## Gene-8 . . 6 2 . 1 2 3
## Gene-9 2 9 . 3 2 . 2 4
## Gene-10 1 1 . . 1 . . 2
## Gene-11 2 9 3 8 . 11 . 1
## Gene-12 4 . 1 2 16 . . 3fake_seurat_2[["RNA"]]$data## 12 x 8 sparse Matrix of class "dgCMatrix"
## Cell-1 Cell-2 Cell-3 Cell-4 Cell-5 Cell-6 Cell-7 Cell-8
## Gene-1 . . 7.014015 . . . 6.034684 6.860015
## Gene-2 6.726633 . 6.321767 . 8.137996 7.071124 6.034684 .
## Gene-3 . 6.726633 7.014015 . . . 6.726633 .
## Gene-4 . . . 7.313887 6.672632 . 8.597420 .
## Gene-5 . . . 6.216606 6.267800 . 6.726633 .
## Gene-6 7.419181 6.034684 7.014015 6.216606 . . . 6.167916
## Gene-7 . 6.726633 6.321767 . 5.576547 7.476306 6.034684 7.775676
## Gene-8 . . 8.112028 6.908755 . 6.378826 6.726633 7.265130
## Gene-9 7.419181 8.229778 . 7.313887 6.267800 . 6.726633 7.552637
## Gene-10 6.726633 6.034684 . . 5.576547 . . 6.860015
## Gene-11 7.419181 8.229778 7.419181 8.294300 . 8.775177 . 6.167916
## Gene-12 8.112028 . 6.321767 6.908755 8.345580 . . 7.265130fake_seurat_2@meta.data## orig.ident nCount_RNA nFeature_RNA nCount_CPM nFeature_CPM ident
## Cell-1 Fake Seurat 12 6 12 6 Fake Seurat
## Cell-2 Fake Seurat 24 6 24 6 Fake Seurat
## Cell-3 Fake Seurat 18 8 18 8 Fake Seurat
## Cell-4 Fake Seurat 20 7 20 7 Fake Seurat
## Cell-5 Fake Seurat 38 7 38 7 Fake Seurat
## Cell-6 Fake Seurat 17 4 17 4 Fake Seurat
## Cell-7 Fake Seurat 24 8 24 8 Fake Seurat
## Cell-8 Fake Seurat 21 8 21 8 Fake SeuratBe sure to examine the converted object to make sure that all of the data you expect actually exist.