1 Linux Basics
1.1 Connecting to the linux server
Open your terminal.
Type the following on your command line, substituting in your username for
.
ssh -p2309 <username>@inbre.ncgr.org- Enter your password.
If you have trouble connecting, please contact Ethan Price at inbre@ncgr.org.
1.2 What are Linux, bash, and the NCGR Server?
A little shell… aka the $ prompt is the command line interface
- A shell is a user interface to the operating system.
- CLI (Command Line Interface)
- GUI (Graphical User Interface)
- Bourne-Again SHell (bash) is a Unix shell and command language
- Each command drives a program or script by talking to the Operating System (Linux)

1.2.1 Find the terminal application you’ll use to log into the NCGR server
For Windows: search for “Tabby” from the Start menu
For Mac: search for “Tabby” using Spotlight (the magnifying glass in the top right of your Mac screen)
1.2.2 Log on to the NCGR server
Enter the following command in the linux terminal to log on to the NCGR Server:
- substitute your personal username in for “username”
ssh -p2309 <username>@inbre.ncgr.orgNotes:
- Because we’re logging in remotely, the -p option is required to specify port 2309.
- If you’re prompted to confirm the connection, say “yes”, then enter your password.
1.3 Linux basics: Part I
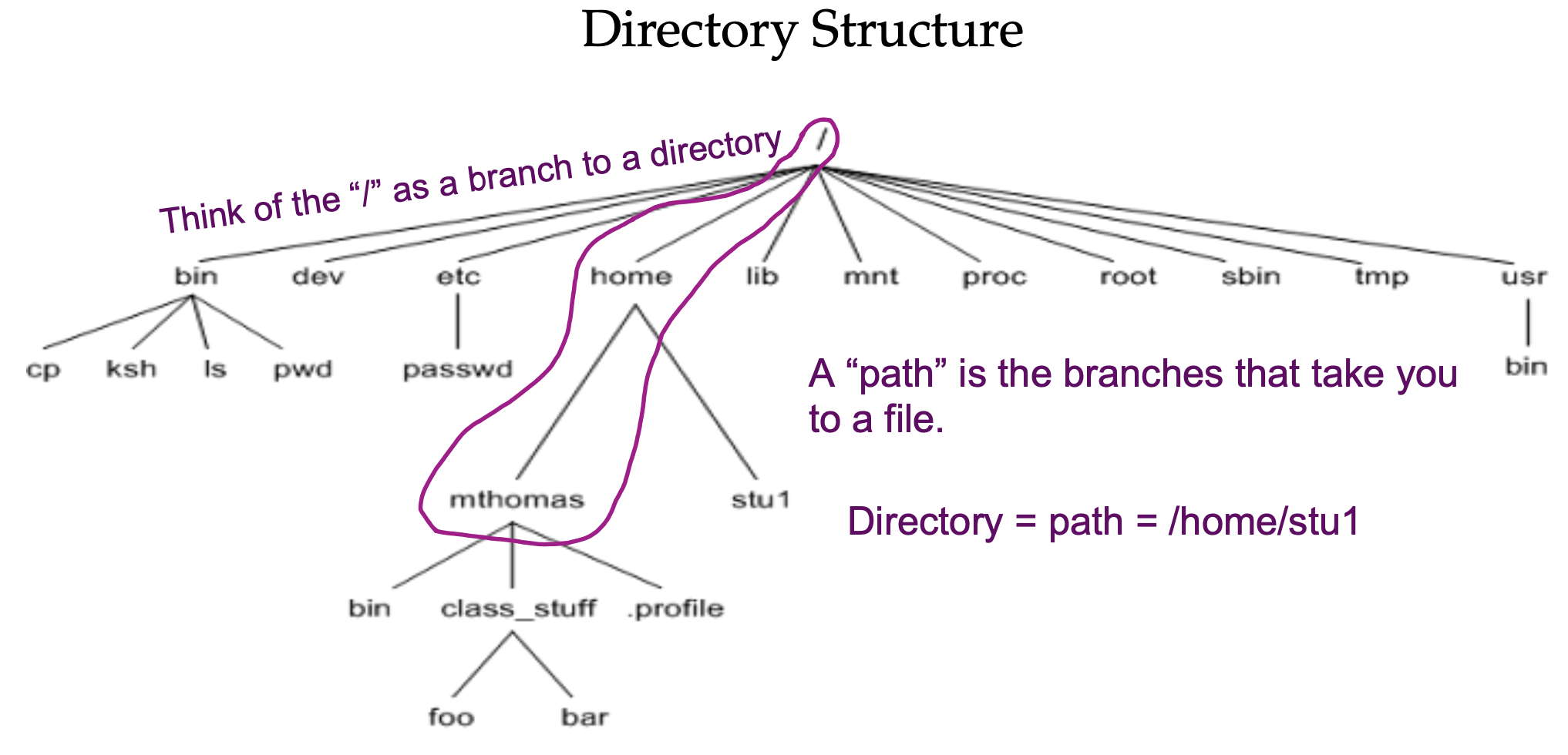
1.3.1 Understanding Directories
print working directory (pwd), mkdir (make directory), and list contents (ls)
pwd## /home/agomez- This is your “home” directory.
Now, create a dir under your home directory for this linux class:
mkdir linuxc
ls## linuxc1.3.2 Listing options
using the ls command
- long list:
ls -l## total 4
## drwx 2 agomez agomez 10 Aug 17 22:50 linuxc- long list, by time, reverse order -old to new:
ls -ltr## total 4
## drwx 2 agomez agomez 10 Aug 17 22:50 linuxc1.3.4 Files: creating with touch command
- Create a file
touch newfile.txt
ls -l## total 0
## -rw-rw-r-- 1 eprice eprice 0 Aug 17 22:50 newfile.txt- Change to your home dir
cd ~
pwd## /home/agomez1.3.5 History command
lists the commands you have entered
history## 17 ls -ltr
## 18 cd ../linuxc
## 19 ls -ltr
## 20 historyScroll through recent commands with the up and down arrows.
To perform a command from the list by number:
!17To perform the last command you made:
!!1.3.6 Files: creating by redirecting standard out
redirect operator >
To send output to a file instead of standard out:
- standard out is just the terminal
history > history.txt
ls -l## total 4
## -rw-rw-r-- 1 eprice eprice 0 Aug 17 22:50 history.txt
## drwxrwxr-x 2 eprice eprice 4096 Aug 17 22:50 linuxccat history.txt## 17 ls -ltr
## 18 cd ../linuxc
## 19 ls -ltr
## 20 history
## 21 ls -ltr
## 20 history > historyNow you have a file with your commands!
1.3.7 File name completion with tab
To autocomplete remainder of file name instead of typing it all in:
- cat h…(press tab)
- cat history
- prevents typos and saves time
1.3.10 Files: securely copying files between your laptop and the NCGR server
secure copy “scp” command
- a secure way to copy files to/from a server while you’re working on an outside network
- like from your home or starbucks to the linux server and vice versa
Syntax: scp [options] sourcepath destinationpath
- Create a file to copy
touch scp_test.txt- Open a local terminal
- do not connect it to the NCGR Server
- Run the scp command from your local terminal window:
- to download the file to your computer from the NCGR Server
- the last period means that the destination is your working directory
scp -P 2309 <username>@inbre.ncgr.org:~/linuxc/scp_test.txt .Note: When you designate a port with secure copy (scp), you use a capital P.
You will be prompted for your NCGR Server password.
Check to see if the file you copied from the NCGR Server is on your computer!
Now upload a file to the NCGR Server from your computer
- Again, run the scp command from your local terminal window:
scp -P 2309 scp_test.txt <username>@inbre.ncgr.org:~/linuxc- Check to see if the file you copied from your computer is on the NCGR Server!
1.3.11 Files and directories: removing files is deleting files
removing “rm” command
Syntax: rm [options] filename
rm -i newfile_bu.txt## rm: remove regular empty file newfile_bu.txt?Enter “yes” or “y” in response to the question:
yes
ls -l## total 0
## -rw-rw-r-- 1 eprice eprice 0 Aug 17 22:50 newfile.txtAt this point everyone should have the above in their linuxc class directory.
1.3.12 Tool box: How to abort a command/process
Hold “control” key then hit “c” key, then release.
- Control-key often referred to as CTRL.
Let’s say you type a command and nothing happens; it hangs. This can happen when the syntax doesn’t make sense. Good time for CTRL c
catIf you can’t execute commands, then CTRL c
## ^CYou should be returned to your prompt: username@single-cell-2309:~/linuxc$
1.4 Linux basics: Part II
1.4.2 Understanding a fasta file format
Fasta files (.fasta or .fa) contain one or more sequences, each preceded by a header starting with “>”.
Show only the first 10 lines of a file with “head” command:
head covid.fasta## >NC_045512.2 |Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co
## ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGATCT
## GTTCTCTAAACGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACT
## CACGCAGTATAATTAATAACTAATTACTGTCGTTGACAGGACACGAGTAACTCGTCTATC
## TTCTGCAGGCTGCTTACGGTTTCGTCCGTGTTGCAGCCGATCATCAGCACATCTAGGTTT
## CGTCCGGGTGTGACCGAAAGGTAAGATGGAGAGCCTTGTCCCTGGTTTCAACGAGAAAAC
## ACACGTCCAACTCAGTTTGCCTGTTTTACAGGTTCGCGACGTGCTCGTACGTGGCTTTGG
## AGACTCCGTGGAGGAGGTCTTATCAGAGGCACGTCAACATCTTAAAGATGGCACTTGTGG
## CTTAGTAGAAGTTGAAAAAGGCGTTTTGCCTCAACTTGAACAGCCCTATGTGTTCATCAA
## ACGTTCGGATGCTCGAACTGCACCTCATGGTCATGTTATGGTTGAGCTGGTAGCAGAACTNow show only the last 10 lines of a file using the “tail” command:
tail covid.fasta## TATTGACGCATACAAAACATTCCCACCAACAGAGCCTAAAAAGGACAAAAAGAAGAAGGC
## TGATGAAACTCAAGCCTTACCGCAGAGACAGAAGAAACAGCAAACTGTGACTCTTCTTCC
## TGCTGCAGATTTGGATGATTTCTCCAAACAATTGCAACAATCCATGAGCAGTGCTGACTC
## AACTCAGGCCTAAACTCATGCAGACCACACAAGGCAGATGGGCTATATAAACGTTTTCGC
## TTTTCCGTTTACGATATATAGTCTACTCTTGTGCAGAATGAATTCTCGTAACTACATAGC
## ACAAGTAGATGTAGTTAACTTTAATCTCACATAGCAATCTTTAATCAGTGTGTAACATTA
## GGGAGGACTTGAAAGAGCCACCACATTTTCACCGAGGCCACGCGGAGTACGATCGAGTGT
## ACAGTGAACAATGCTAGGGAGAGCTGCCTATATGGAAGAGCCCTAATGTGTAAAATTAAT
## TTTAGTAGTGCTATCCCCATGTGATTTTAATAGCTTCTTAGGAGAATGACAAAAAAAAAA
## AAAAAAAAAAAAAAAAAAAAAAAThe pipe operator will redirect output of a command to another command.
Use the pipe operator to redirect “cat” output to “head”:
- The symbol “|” denotes a pipe
cat covid.fasta | head## >NC_045512.2 |Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co
## ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGATCT
## GTTCTCTAAACGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACT
## CACGCAGTATAATTAATAACTAATTACTGTCGTTGACAGGACACGAGTAACTCGTCTATC
## TTCTGCAGGCTGCTTACGGTTTCGTCCGTGTTGCAGCCGATCATCAGCACATCTAGGTTT
## CGTCCGGGTGTGACCGAAAGGTAAGATGGAGAGCCTTGTCCCTGGTTTCAACGAGAAAAC
## ACACGTCCAACTCAGTTTGCCTGTTTTACAGGTTCGCGACGTGCTCGTACGTGGCTTTGG
## AGACTCCGTGGAGGAGGTCTTATCAGAGGCACGTCAACATCTTAAAGATGGCACTTGTGG
## CTTAGTAGAAGTTGAAAAAGGCGTTTTGCCTCAACTTGAACAGCCCTATGTGTTCATCAA
## ACGTTCGGATGCTCGAACTGCACCTCATGGTCATGTTATGGTTGAGCTGGTAGCAGAACTcat covid.fasta | tail## TATTGACGCATACAAAACATTCCCACCAACAGAGCCTAAAAAGGACAAAAAGAAGAAGGC
## TGATGAAACTCAAGCCTTACCGCAGAGACAGAAGAAACAGCAAACTGTGACTCTTCTTCC
## TGCTGCAGATTTGGATGATTTCTCCAAACAATTGCAACAATCCATGAGCAGTGCTGACTC
## AACTCAGGCCTAAACTCATGCAGACCACACAAGGCAGATGGGCTATATAAACGTTTTCGC
## TTTTCCGTTTACGATATATAGTCTACTCTTGTGCAGAATGAATTCTCGTAACTACATAGC
## ACAAGTAGATGTAGTTAACTTTAATCTCACATAGCAATCTTTAATCAGTGTGTAACATTA
## GGGAGGACTTGAAAGAGCCACCACATTTTCACCGAGGCCACGCGGAGTACGATCGAGTGT
## ACAGTGAACAATGCTAGGGAGAGCTGCCTATATGGAAGAGCCCTAATGTGTAAAATTAAT
## TTTAGTAGTGCTATCCCCATGTGATTTTAATAGCTTCTTAGGAGAATGACAAAAAAAAAA
## AAAAAAAAAAAAAAAAAAAAAAA1.4.3 Understanding fastq (fq) file format
Fastq files contain sequence reads and associated meta data.
ln -s /home/data/single-cell-2309/SP1.fq
ls -ltr## total 0
## -rw-rw-r-- 1 eprice eprice 0 Aug 17 22:50 newfile.txt
## lrwxrwxrwx 1 eprice eprice 26 Aug 17 22:50 covid.fasta -> /home/eprice/covid.fasta
## lrwxrwxrwx 1 eprice eprice Aug 17 22:50 SP1.fq -> /home/fds/unix_basics/SP1.fqTail the last 4 lines:
tail -n 4 SP1.fq## @cluster_834:UMI_TTAAGG
## AGGGTGGGGGATCACATTTATTGTATTGAGG
## +
## =A=@AB===>4?A=??EEB?EB@C?ECB=A?A fastq file has 4 lines per record:
- The header; starts with “@”
- The sequence
- Throwaway line; begins with “+”
- Phred-scaled quality scores
1.4.4 Using grep (global regular expression print) to extract metrics
Grep will output the lines containing a provided expression.
Syntax: grep [options] “expression” filename
grep ">" covid.fasta## >NC_045512.2 |Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co
## >MT627325.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT622319.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT568634.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT568635.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT568636.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT568637.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT568638.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT568639.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT568640.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT568641.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407649.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407650.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407651.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407652.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407653.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407654.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407655.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407656.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407657.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407658.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT407659.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT534630.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT510727.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT510728.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079843.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079844.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079845.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079846.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079847.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079848.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079849.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079850.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079851.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079852.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079853.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT079854.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT446312.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT412134.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT396241.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT039874.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT281577.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291826.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291827.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291828.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291829.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291830.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291831.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291832.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291833.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291834.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291835.2 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT291836.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT259226.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT259227.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT259228.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT259229.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT259230.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT259231.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253696.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253697.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253698.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253699.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253700.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253701.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253702.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253703.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253704.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253705.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253706.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253707.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253708.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253709.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT253710.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT226610.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT121215.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT135041.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT135042.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT135043.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT135044.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT123290.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT123291.2 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT123292.2 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT123293.2 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT093631.2 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT049951.1 |Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/huma
## >MT039873.1 |Severe acute respiratory syndrome coronavirus 2 isolate HZ-1, complete
## >MT019529.1 |Severe acute respiratory syndrome coronavirus 2 isolate BetaCoV/Wuhan/I
## >MT019530.1 |Severe acute respiratory syndrome coronavirus 2 isolate BetaCoV/Wuhan/I
## >MT019531.1 |Severe acute respiratory syndrome coronavirus 2 isolate BetaCoV/Wuhan/I
## >MT019532.1 |Severe acute respiratory syndrome coronavirus 2 isolate BetaCoV/Wuhan/I
## >MT019533.1 |Severe acute respiratory syndrome coronavirus 2 isolate BetaCoV/Wuhan/I
## >MN996527.1 |Severe acute respiratory syndrome coronavirus 2 isolate WIV02, complete
## >MN996528.1 |Severe acute respiratory syndrome coronavirus 2 isolate WIV04, complete
## >MN996529.1 |Severe acute respiratory syndrome coronavirus 2 isolate WIV05, complete
## >MN996530.1 |Severe acute respiratory syndrome coronavirus 2 isolate WIV06, complete
## >MN996531.1 |Severe acute respiratory syndrome coronavirus 2 isolate WIV07, complete
## >MN988668.1 |Severe acute respiratory syndrome coronavirus 2 isolate 2019-nCoV WHU01
## >MN988669.1 |Severe acute respiratory syndrome coronavirus 2 isolate 2019-nCoV WHU02
## >MN938384.1 |Severe acute respiratory syndrome coronavirus 2 isolate 2019-nCoV_HKU-S
## >MN975262.1 |Severe acute respiratory syndrome coronavirus 2 isolate 2019-nCoV_HKU-S
## >MN908947.3 |Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, comAdding the “-c” option counts the number of lines containing a match
- not the number of matches!
grep -c ">" covid.fasta## 102The “-v” option reverses the grep search, which is the first step towards finding the total length of sequences
grep -v ">" covid.fastaUse
We can add in the pipe operator to redirect our output to the “wc” command.
The output shows the number of newline characters, followed by line count and total character count.
grep -v ">" covid.fasta | wcwc: prints newline, word, and byte counts for each file:
## 50812 50812 3096018Let’s trim out the newline characters with “tr -d” before doing the word count:
grep -v ">" covid.fasta | tr -d '\n' | wc## 0 1 30452061.4.5 Working with compressed files
Powerful Z commands (zcat)
- p. 1 of 2
Let’s copy another file:
cp /home/data/single-cell-2309/table1.txt.gz .
zcat table1.txt.gz
## 1, Justin Timberlake, Title 545, Price $7.30
## 2, Taylor Swift, Title 723, Price $7.90
## 3, Mick Jagger, Title 610, Price $7.90
## 4, Lady Gaga, Title 118, Price $7.30
## 5, Johnny Cash, Title 482, Price $6.50
## 6, Elvis Presley, Title 335, Price $7.30
## 7, John Lennon, Title 271, Price $7.90
## 8, Michael Jackson, Title 373, Price $5.50zgrep "Jagger" table1.txt.gz## 3, Mick Jagger, Title 610, Price $7.90!! = last command
:s = substitute /word1/with word2
!!:s/Jagger/John## 5, Johnny Cash, Title 482, Price $6.50
## 7, John Lennon, Title 271, Price $7.901.4.6 Start ^ and end $ symbols
Powerful Z commands (zgrep)
- p. 2 of 2
Display all the lines that start with 8:
zgrep "^8" table1.txt.gz## 8, Michael Jackson, Title 373, Price $5.50Display all the lines that end with 50:
zgrep "50$" table1.txt.gz## 5, Johnny Cash, Title 482, Price $6.50
## 8, Michael Jackson, Title 373, Price $5.501.4.7 Files: parsing and creating data-subsets
p.1 of 5
Be sure you are in linuxc directory (as usual)
AWK command
- Aho, Weinberger and Kernighan
- the authors of the language
General syntax:
- awk ’pattern {action}’ input-file
- output goes to standard out=terminal
- awk ’pattern {action}’ input-file > output-file
- or send to an output file
1.4.8 Files: parsing and creating data-subsets
p. 2 of 5
gunzip table1.txt.gz
cat table1.txt## 1, Justin Timberlake, Title 545, Price $7.30
## 2, Taylor Swift, Title 723, Price $7.90
## 3, Mick Jagger, Title 610, Price $7.90
## 4, Lady Gaga, Title 118, Price $7.30
## 5, Johnny Cash, Title 482, Price $6.50
## 6, Elvis Presley, Title 335, Price $7.30
## 7, John Lennon, Title 271, Price $7.90
## 8, Michael Jackson, Title 373, Price $5.50awk '{print $1 $3 $5}' table1.txt## 1,Timberlake,545,
## 2,Swift,723,
## 3,Jagger,610,
## 4,Gaga,118,
## 5,Cash,482,
## 6,Presley,335,
## 7,Lennon,271,
## 8,Jackson,373,1.4.9 Files: parsing and creating data-subsets
p. 3 of 5
Using field separator command -F
cat table1.txt## 1, Justin Timberlake, Title 545, Price $7.30
## 2, Taylor Swift, Title 723, Price $7.90
## 3, Mick Jagger, Title 610, Price $7.90
## 4, Lady Gaga, Title 118, Price $7.30
## 5, Johnny Cash, Title 482, Price $6.50
## 6, Elvis Presley, Title 335, Price $7.30
## 7, John Lennon, Title 271, Price $7.90
## 8, Michael Jackson, Title 373, Price $5.50awk -F, '{print $3}' table1.txt## Title 545
## Title 723
## Title 610
## Title 118
## Title 482
## Title 335
## Title 271
## Title 3731.4.10 Files: parsing and creating data-subsets
p. 4 of 5: Conditional awk
Time to really watch for syntax errors!
Statements inside the curly brackets {statement} are called a block.
If you put a conditional expression in front of a block with with ==, the statement inside the block will be executed only if the condition is true.
The whole awk command is inside ’ ’.
- For example:
- awk ‘$7==“$7.30”{print $3}’ table1.txt
- The condition is:
- if $7==“$7.30”
- meaning if the element at column 7 is equal to “$7.30”, then execute statement(s) in the block {print $3}.
1.4.11 Revisiting table1 and previous awk command
p. 5 of 5
awk '$7=="$7.30" {print $3}' table1.txt## Timberlake,
## Gaga,
## Presley,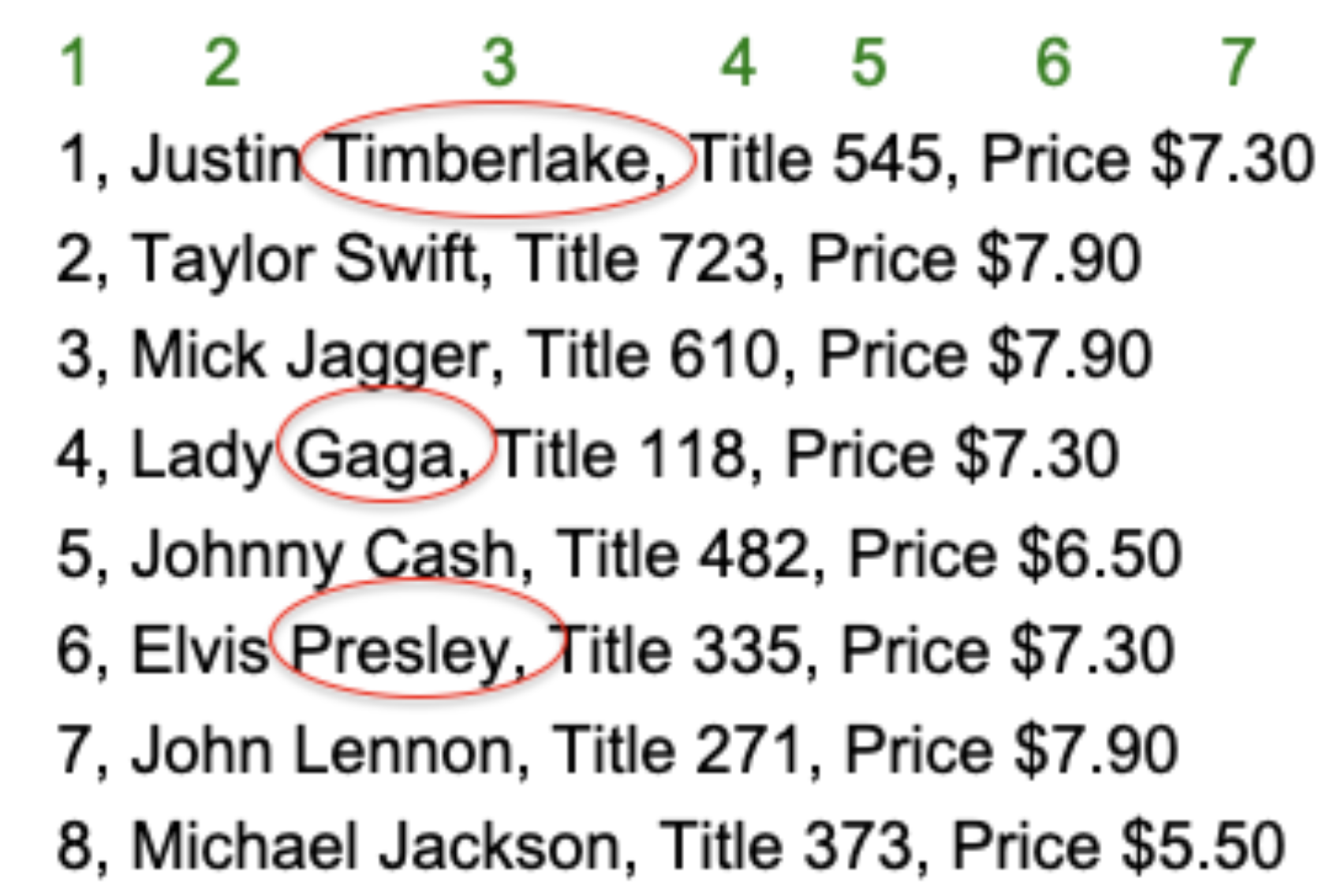
1.4.12 Files: Stream EDitor (sed)
text substitution
Syntax: sed s/pattern/replacement
Examples:
echo "it’s a trap" | sed s/ra/ar/## it’s a tarpSay you want to change all price occurrences of $7.90 to $8.90 from table1.txt, and save the changes to a new file.
You can do this with sed.
sed 's/7.90/8.90/' table1.txt > table2.txtUse cat to display the contens of the new file:
cat table2.txt## 1, Justin Timberlake, Title 545, Price $7.30
## 2, Taylor Swift, Title 723, Price $8.90
## 3, Mick Jagger, Title 610, Price $8.90
## 4, Lady Gaga, Title 118, Price $7.30
## 5, Johnny Cash, Title 482, Price $6.50
## 6, Elvis Presley, Title 335, Price $7.30
## 7, John Lennon, Title 271, Price $8.90
## 8, Michael Jackson, Title 373, Price $5.501.4.13 The Bash “for” Loop
Suppose we want to run a command for a group of files in a directory. We can use a for loop to target all of them at once.
Syntax: for variablename in filenameexpression; do command ${variablename}; done
for file in *; do echo ${file}; done## covid.fasta
## newfile.txt
## SP1.fq
## table1.txt
## table2.txt- The part up to the first semicolon targets every file in the working directory with the “*” wildcard
- The second part will sequentially echo each file in the working directory
- The third part is required to terminate the loop
1.4.14 Help with command syntax
If you forget details of a certain command, documentation can easily be found with a web search.
There is also cheat sheet on the weebly site under Supplemental Documents.
1.4.15 Exercises
Using awk, print to output the first names of artists with album prices over $7.50 from ta- ble1.txt. Then redirect this output to a file named homework_1.txt
Using sed, replace all commas with semicolons in table1.txt. Save this to a file named home- work_2.txt
Piping history to grep, show all commands you’ve used with the expression “ls”. Save this to a file named homework_3.txt
Piping cat to wc -l on the history text file made during the tutorial, count the number of lines in it.
Using scp, download table1.txt to your own machine. Check it’s there, then upload it back to the NCGR server.
1.5 Linux basics: Part III
Exercise
Log on to the NCGR Server
Enter the following command to log on to the NCGR Server:
ssh -p2309 <username>@inbre.ncgr.org- Don’t forget to substitute your personal username in
- Make a new directory under your home directory:
mkdir fastq_files- Enter into the new directory:
cd fastq_files- Move the fastq file from yesterday to the present working directory:
mv ~/linuxc/SP1.fq .
ls -ltr## total 0
## lrwxrwxrwx 1 elavelle elavelle 28 Aug 17 22:50 SP1.fq -> /home/data/metagenomics-2310/SP1.fq- How can we count the number of records in a fastq file?
grep -c "@cluster" SP1.fq## 250- If you want to determine the number of lines in a file, you can use the “wc” command.
cat SP1.fq | wc -l## 1000- Why does the first command output 250 and the second 1000?
1.5.1 More Exercises
With one command, send a copy of table1.txt in the linuxc directory to your home directory with the name table1_bu.txt
Print to standard output the last line of table1.txt
Use a loop to count the number of lines in all files in the linuxc directory.
Print to standard output the last names of music artists with album prices less than or equal to $7.30
Create a file with only the accession numbers of the sequences contained in the covid.fasta file (with no additional spaces or symbols).