Chapter 4 Community Profiling Part II
4.1 Structural and Functional Approaches to study microbiomes
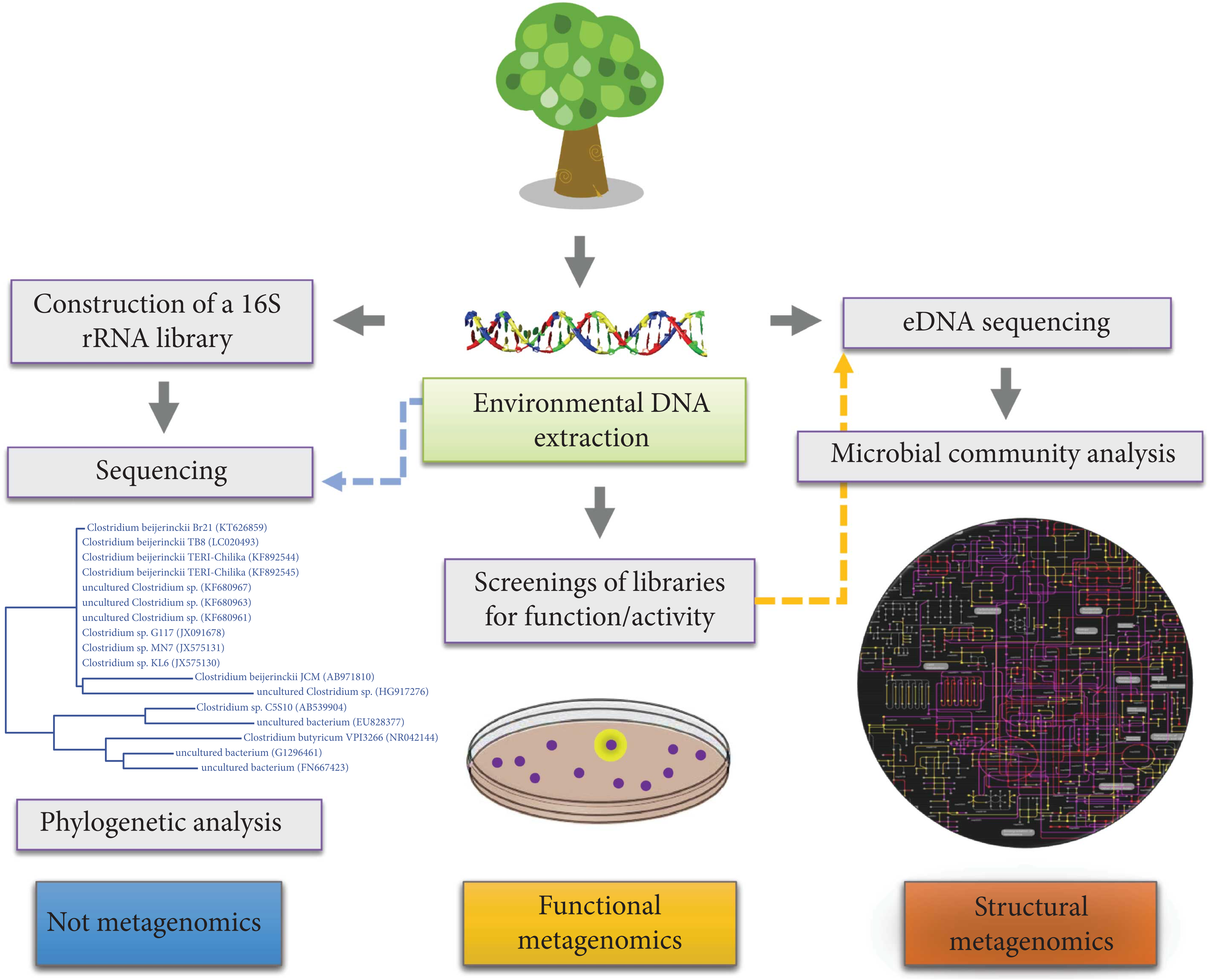
4.1.1 16S rRNA as an evolutionary chronometer
- Ubiquitous – present in all known life (excluding viruses)
- Functionally constant wrt translation and secondary structure
- Evolves very slowly – mutations are extremely rare
- Large enough to extract information for evolutionary inference
- Limited exchange – limited examples of rRNA gene sharing between organisms
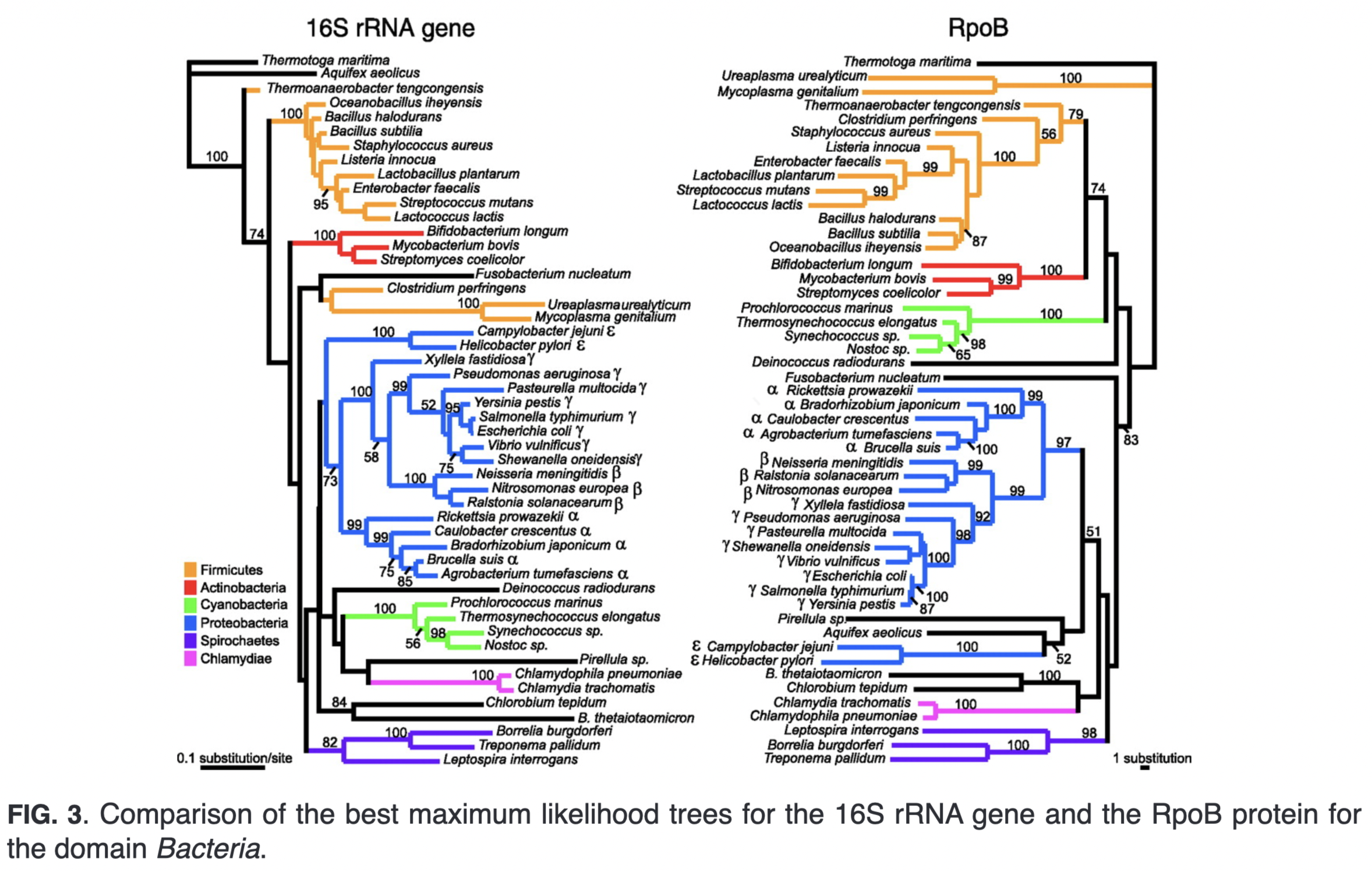
4.1.2 16S rRNA vs rpoB (RNA polymerase β subunit gene)
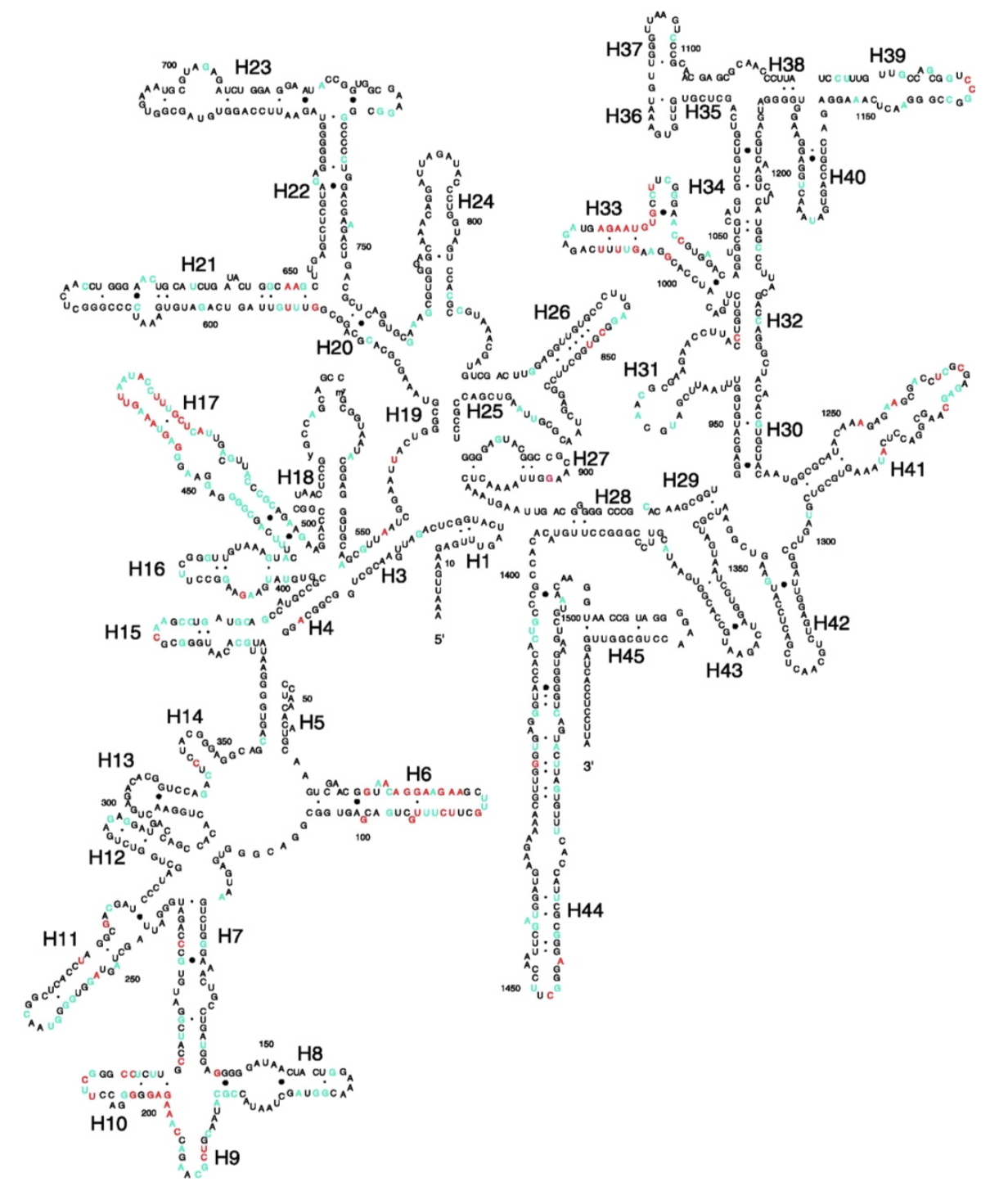
4.1.2.1 16S rRNA hypervariable regions

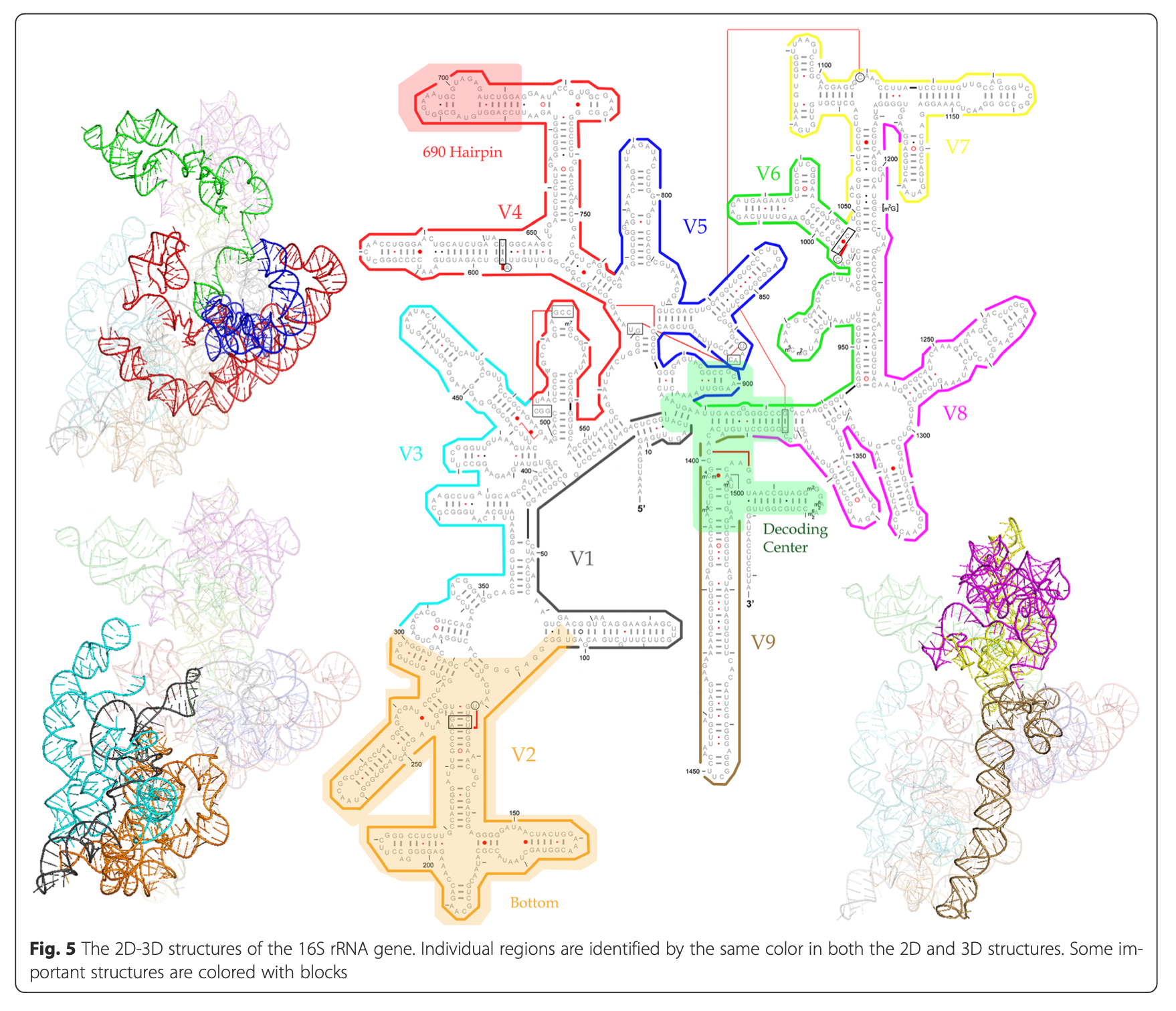
Illustration of different hypervariable regions of 16S rRNA
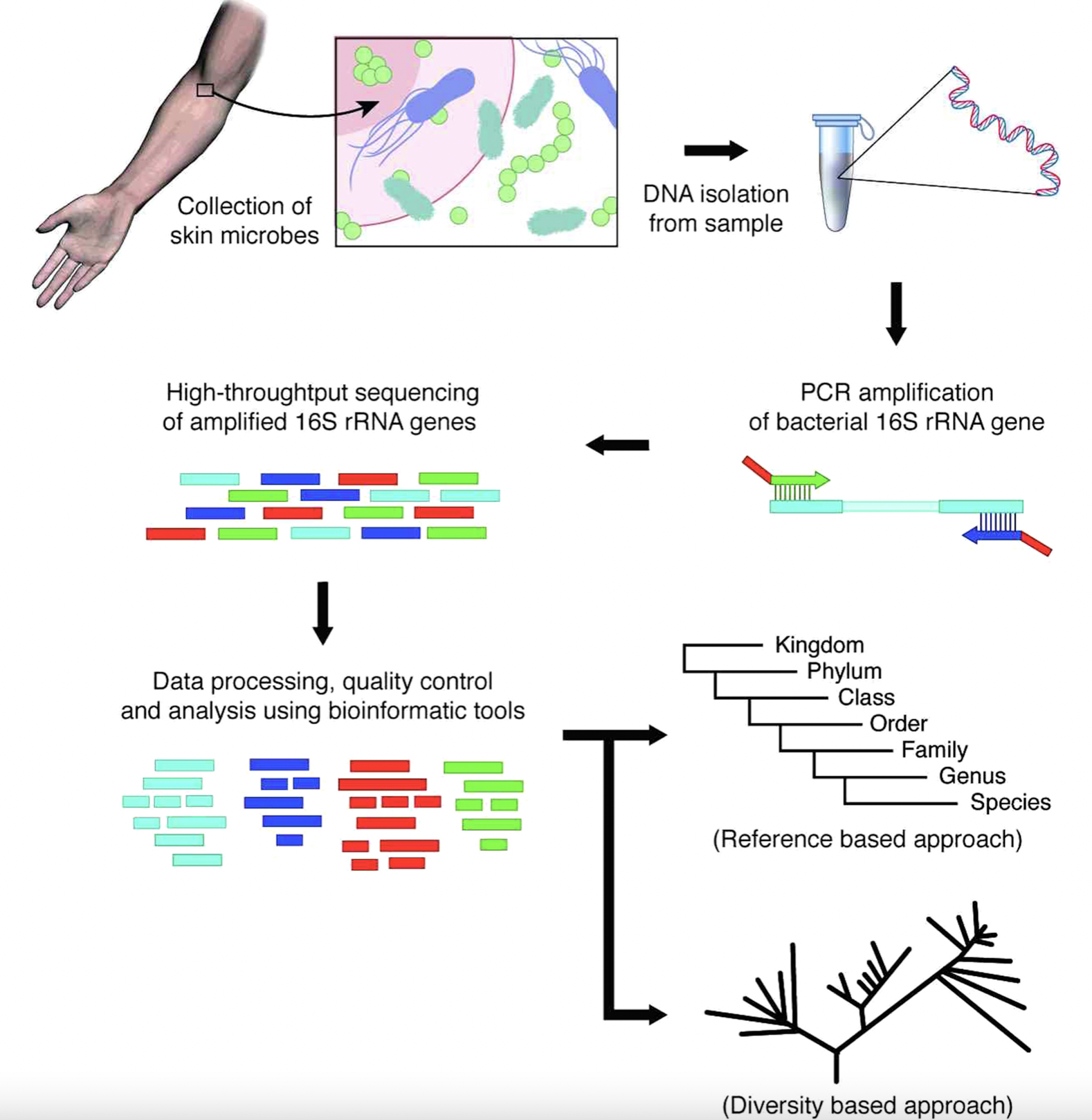
4.2 Basic Workflow for 16S Gene Based Sequencing
4.3 Addressing the ‘fine print’ while generating 16S rRNA gene amplicon libraries
- Sample Collection
- Sample collection significantly influences the microbiome profiler after sequencing
- Sample storage
- DNA isolation
- Template concentration
- Template extraction protocol
- PCR amplification
- PCR bias and inhibitors
- Amplification of contaminants
J. Microbiol Methods (2018), App. Environ. Microbiol. (2014), Microbiome (2015)
4.4 Steps Involved
- Experimental Design: How many samples can be included in the sequencing run?
- By using barcoded primers, numerous samples can be sequenced simultaneously (multiplexing)
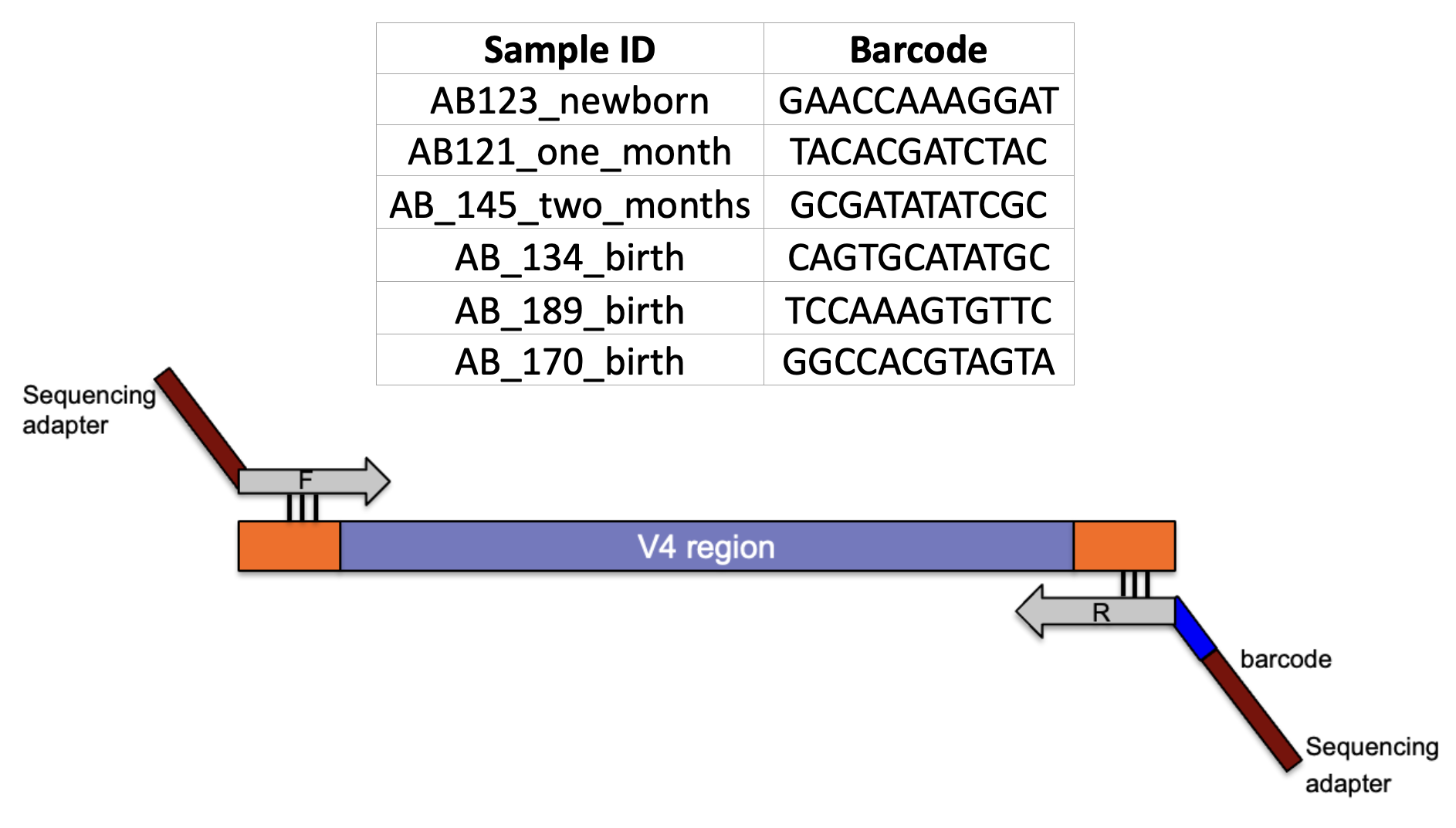
4.4.1 Samples
- More the number of samples, more cost effective the run (sequencing depth will be compromised)
Comparison of multiplexing capacity by sequencing system
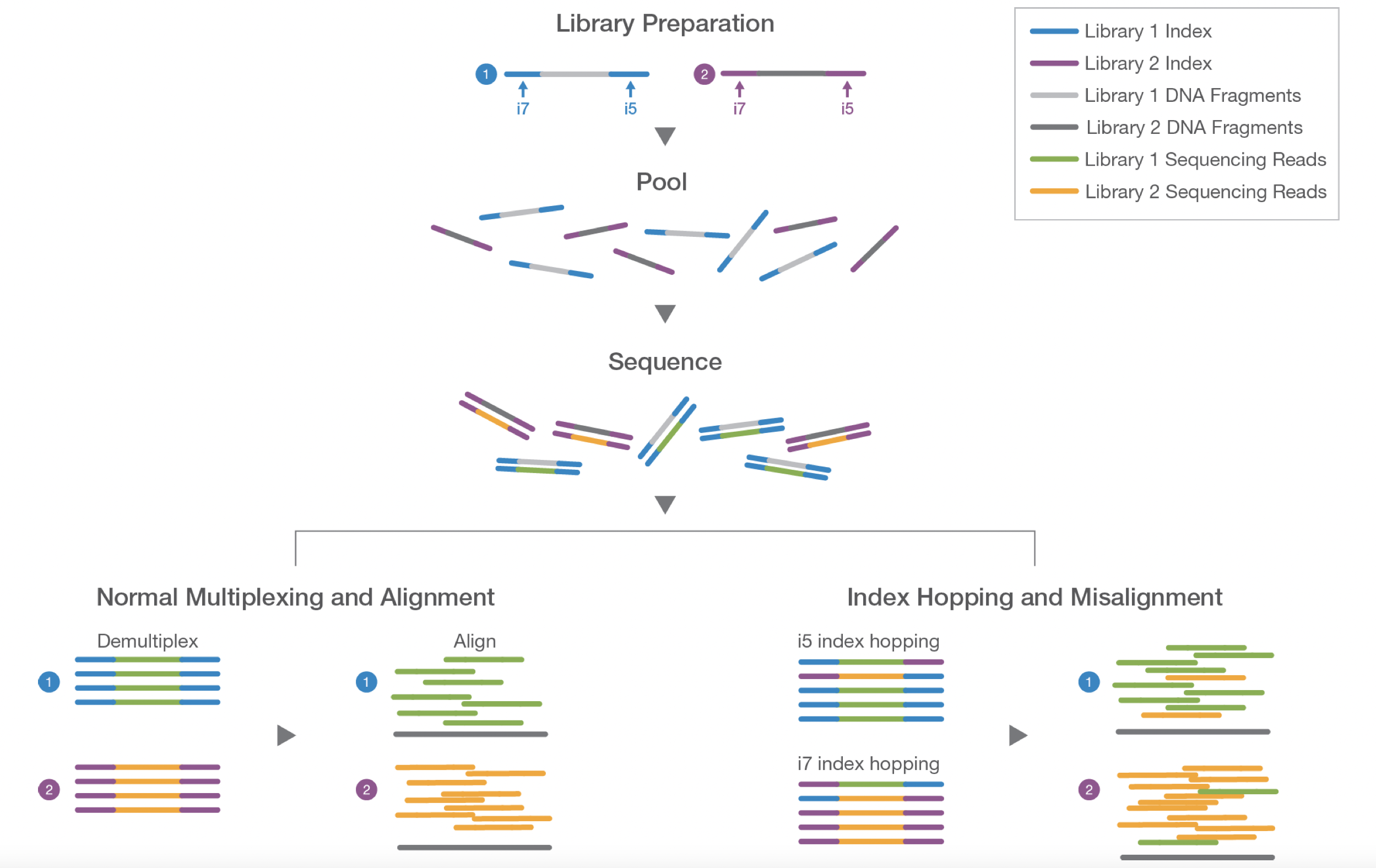
- It is critical to have a ‘library prep manifest’ to document the position of each sample with its associated barcode along with additional metadata information
4.4.2 Include Controls
- Between run repeat (process any sample in duplicate per run to measure reproducibility across runs)
- Within run repeat (process any sample in duplicate per plate to measure reproducibility)
- Water used during PCR (water blank- to determine if any contaminant was introduced during PCR reaction)
- Water spiked with known bacterial DNA (mock bacterial communities- enables quantification of sequencing errors, minimizes bias during sampling and library preparation )
4.4.3 DNA extraction protocol
- Effect of mechanical lysis methods for extraction
- Presence of inhibitors such as organic matter, humic acid, bile salts, polysaccharides
- DNA yield post extraction and reproducibility
Effect of bead beating was larger than sampling time over 5 months
The effect of bead beating on the observed microbial community composition:
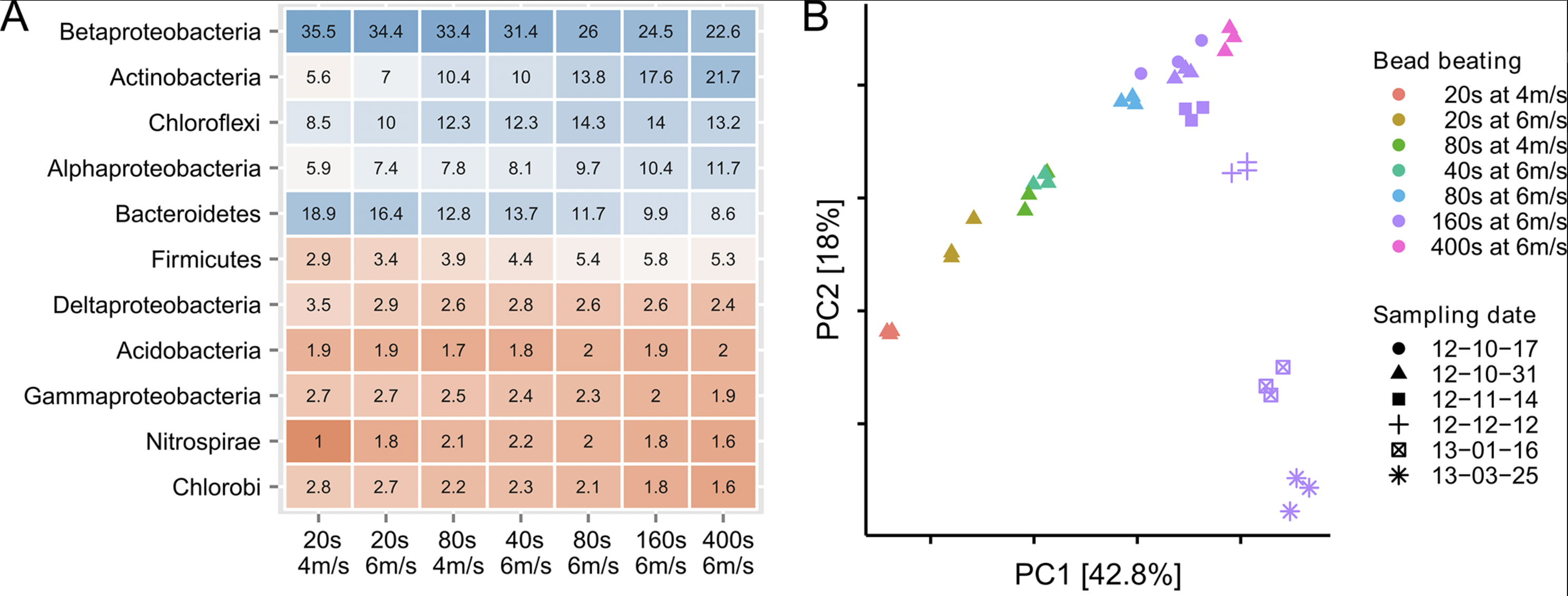
A. Percentage read abundance of the 11 most abundant phyla as a result of bead beating intensity
B. PCA of samples with different bead beating intensities vs. samples taken at different dates
4.4.4 Selection of primers and region of 16S gene influence microbial profile
V2, V4, V6-V7 regions produced consistent results

- V2, V3 and V6 contain maximum nucleotide heterogeneity
- V6 is the shortest hypervariable region with the maximum sequence heterogeneity
- V1 is best target for distinguishing pathogenic S aureus
- V2 and V3 are excellent targets for speciation among Staph and Strep pathogens as well as Clostridium and Neisseria species
- V2 especially useful for speciation of Mycobacterium sp. and detection of E coli O157:H7
- V3 useful for speciation of Haemophilus sp
- V6 best target for probe based PCR assays to identify CDC select agents (bio-terrorism agents)

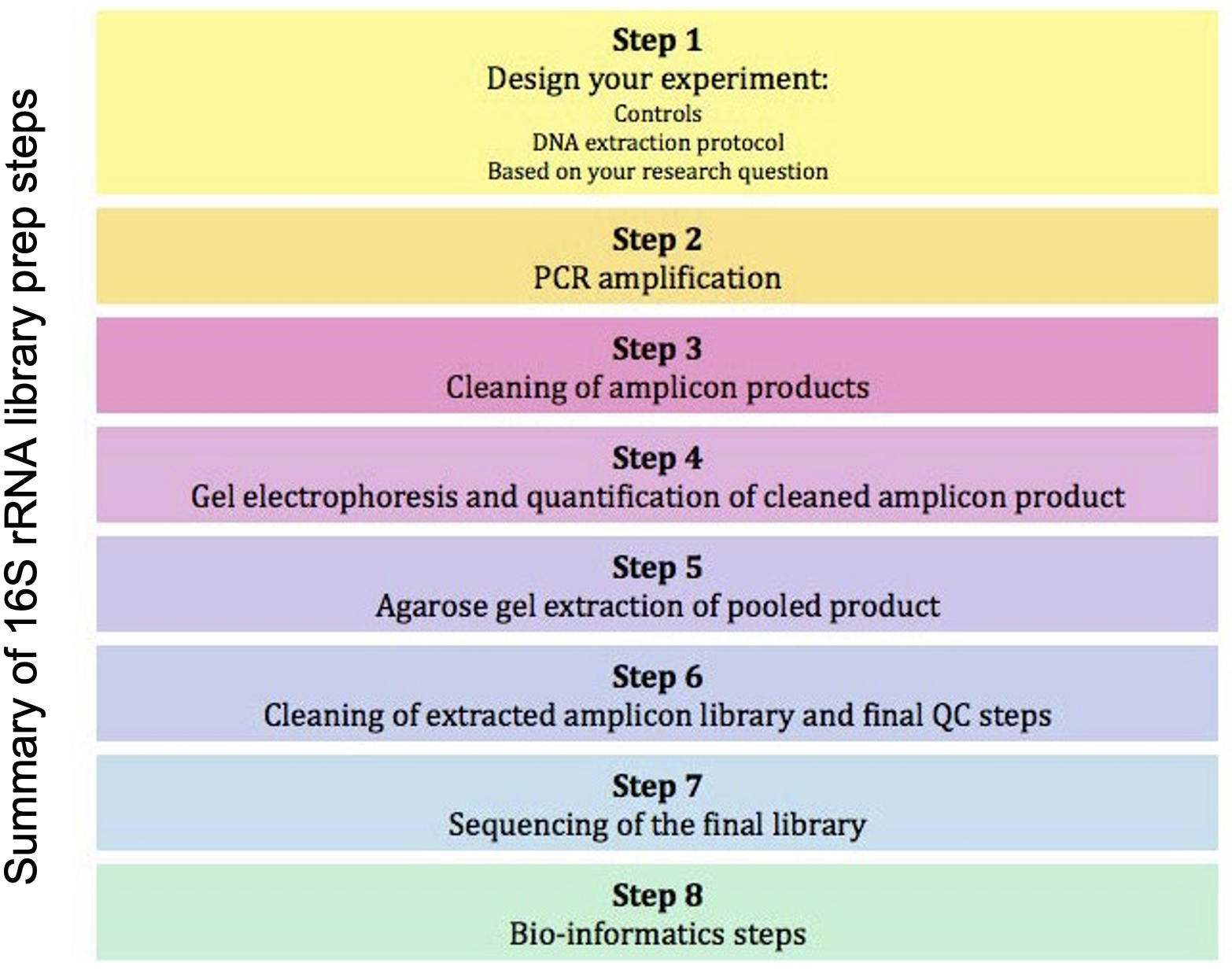
4.4.5 Purification of Amplicons
After one –step or two-step PCR, products are cleaned up using AMpure beads
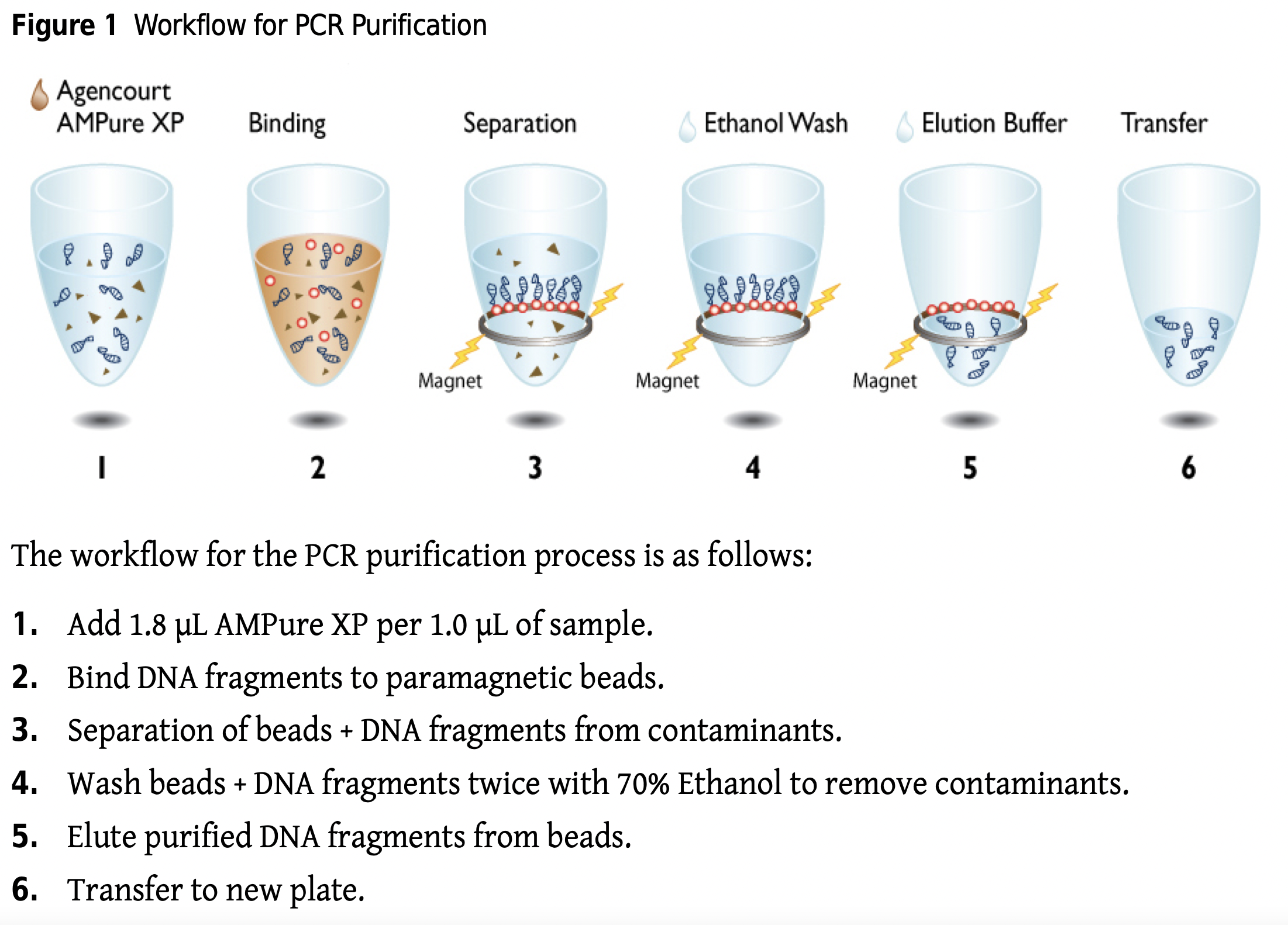
- Gel Electrophoresis and quantification of cleaned amplicon products
- Qubit
- Sample pooling – equimolar concentrations (how many samples do you want to pool? How many reads per sample?
- Gel extraction of pooled product
- Final clean up (Qiagen kit) and QC
4.5 Oxford Nanopore Sequencing
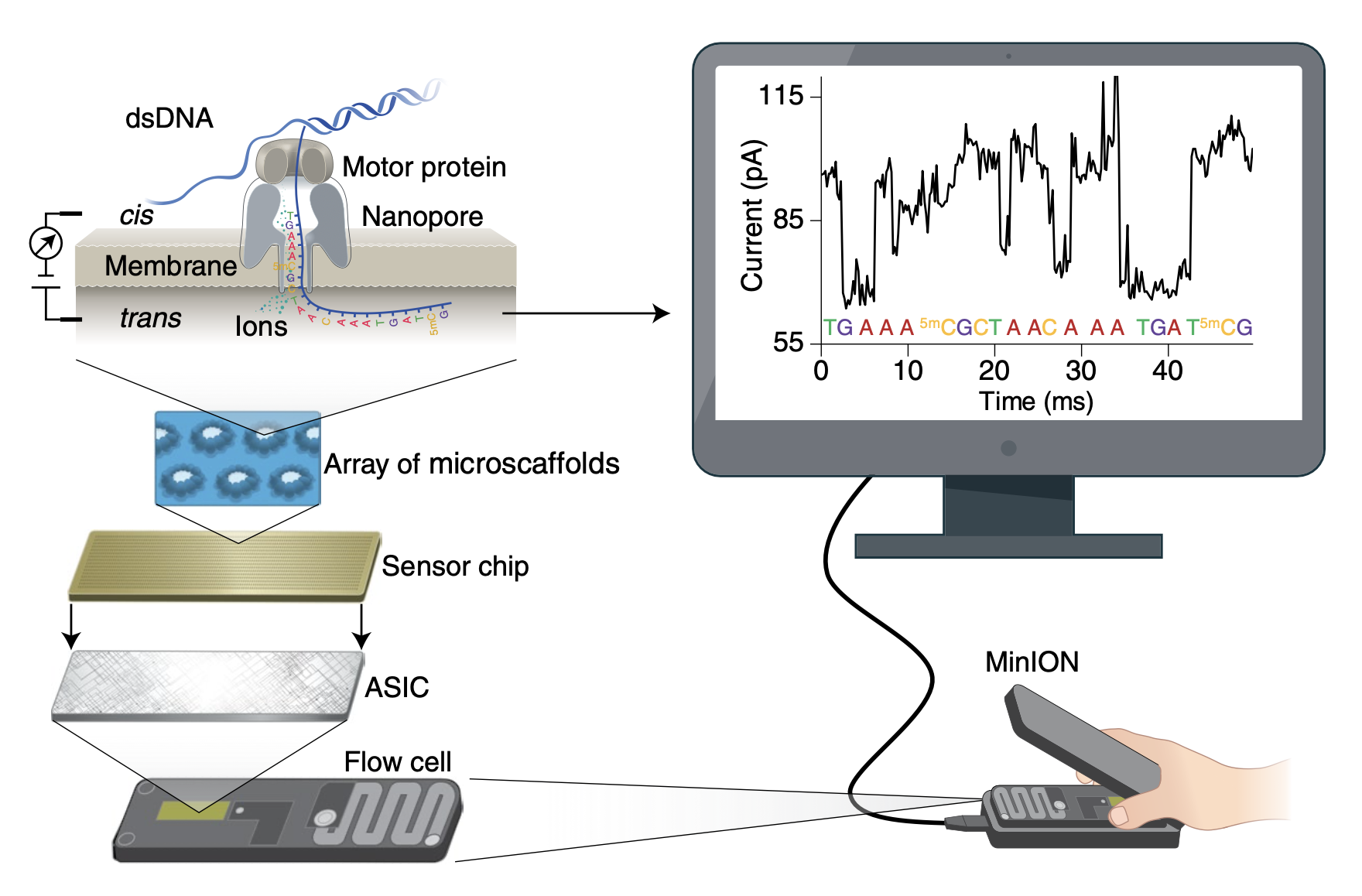
4.5.1 How does it work?
- Proetin pore
- nanoscale
- biosensor
- motor protein ratchets DNA/RNA through
- Ionic current
- constant voltage
- in electrolytic solution
- disrupted by nucleotide sequence
- changes in current correspond to sequence
Watch the video below and answer questions:
https://www.youtube.com/watch?v=RcP85JHLmnI
- How does the DNA bind to the pore?
- Does something help guide the DNA to the pore?
- What is the signal produced by the DNA?
https://www.youtube.com/watch?v=E9-Rm5AoZGw
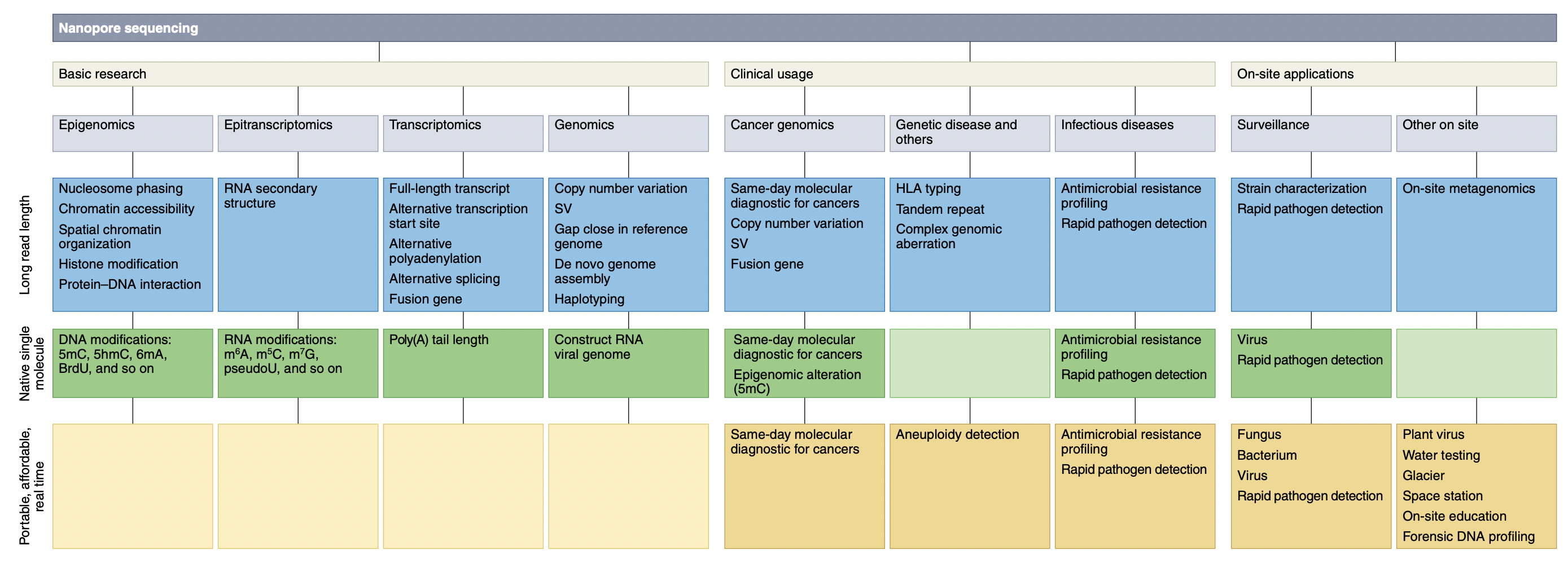
Applications for ONT sequencing:
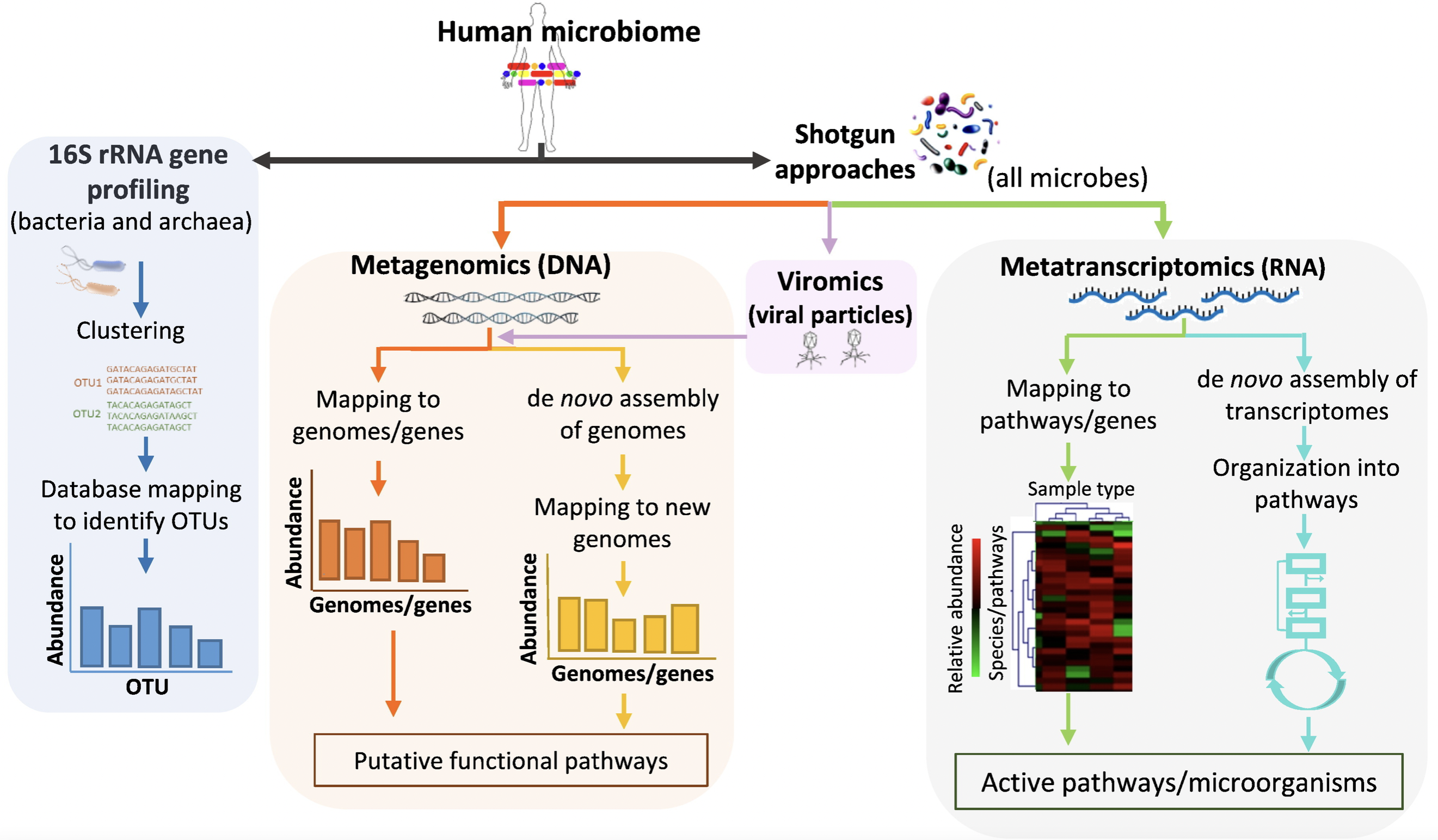
4.6 Metagenomics and Metatranscriptomics
Metagenomics: Untargeted sequencing of all microbial genomes present in a sample.
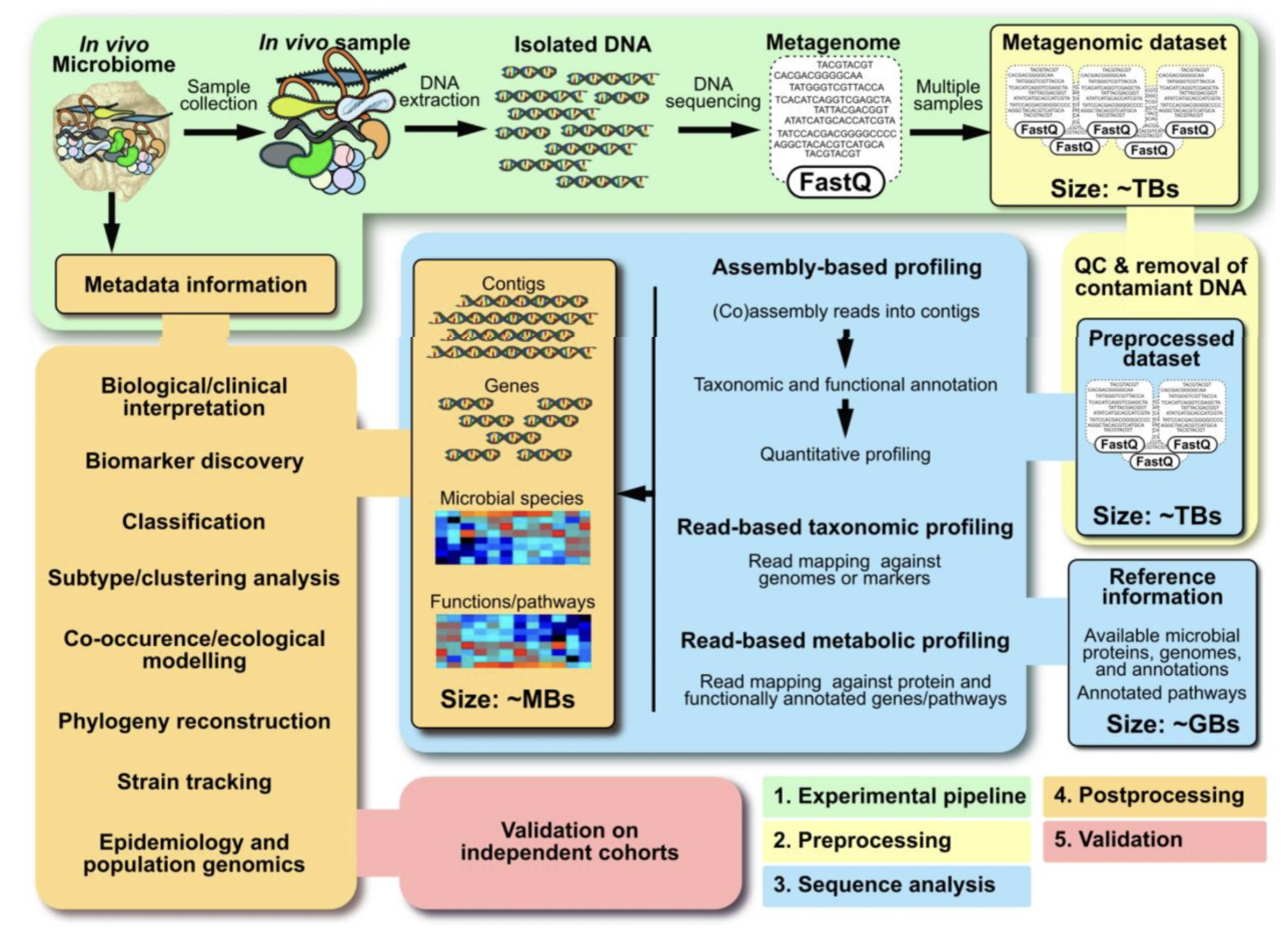
4.6.1 Shotgun Metagenomics
- Study design and experimental protocol
- Computational pre-processing
- Sequence analysis
- Post-processing
- Validation
4.6.2 Sample collection and DNA extraction
- Sample collection and preservation methods can affect quality and accuracy of metagenomic data
- Collect sufficient biomass
- Minimize contamination
- Enrichment methods where applicable
- DNA extraction methods can affect the composition of downstream sequence data
- Method must be effective for diverse microbial taxa
- Mechanical lysis (bead beating) method is considered superior, however, data will be biased for easy-to-lyse microbes
- Bead beating will result in short DNA fragments and lead to DNA loss during library prep methods.
4.6.3 Sources of contamination
- Kit or lab reagents
- Low biomass samples are vulnerable to contamination as there is less ‘real’ signal to compete with low levels of contamination
- Use ultraclean kits
- Include blank sequencing controls
- Cross- over from previous sequencing runs
- PhiX control DNA
- Human/ host DNA
4.6.4 Coverage and Sequencing considerations
- No published guidelines for ‘correct’ amount of coverage for a given environment
Choose a system that maximizes output in order to recover sequences from as many low-abundance members of the microbiome as possible
HiSeq 2500 or 4000, NextSeq and NovaSeq produce high volume data (120Gb- 1.5 Tb per run) – suited for metagenomics study
Multiplexing prudently will enable desired per-sample sequencing depth
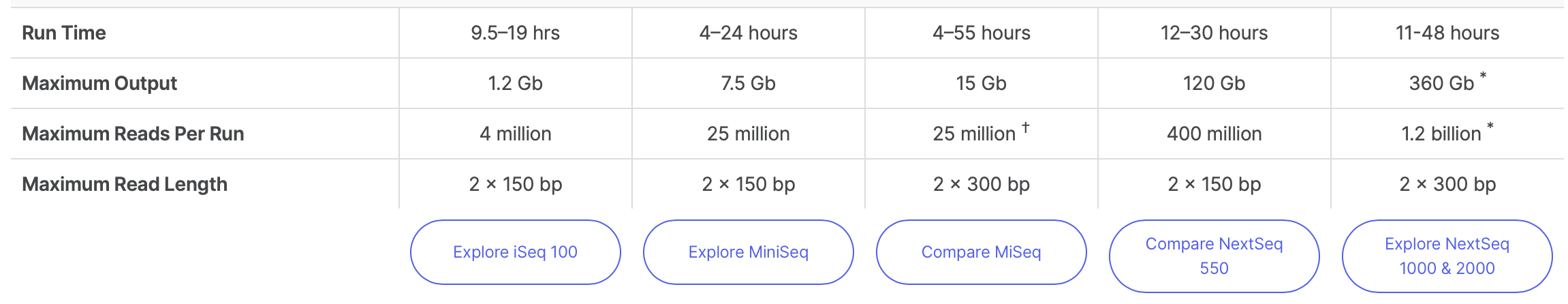
4.6.5 Illumina Sequencers and Yield
https://www.illumina.com/systems/sequencing-platforms.html
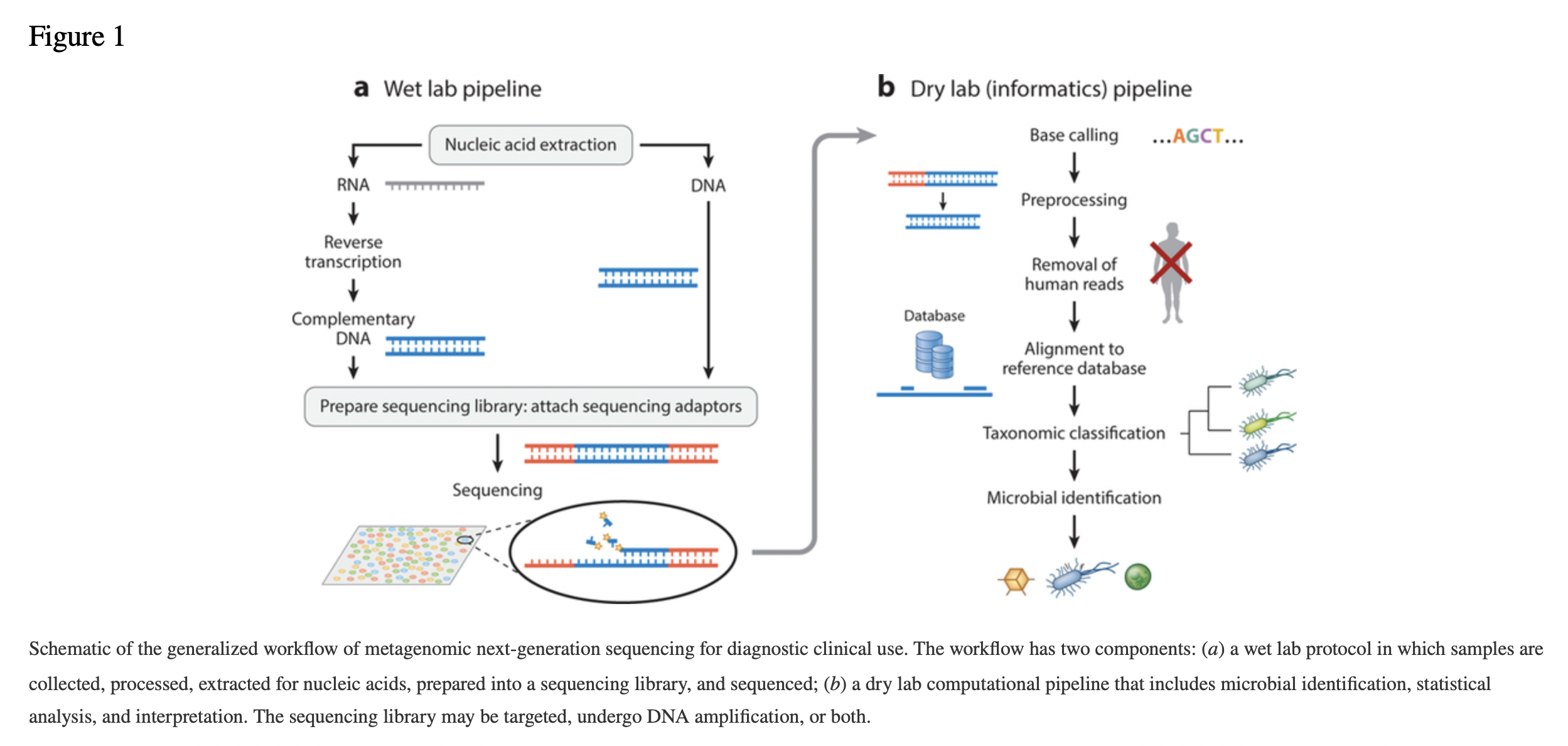
4.6.6 Generalized workflow of metagenomic next-generation sequencing for diagnostic clinical use
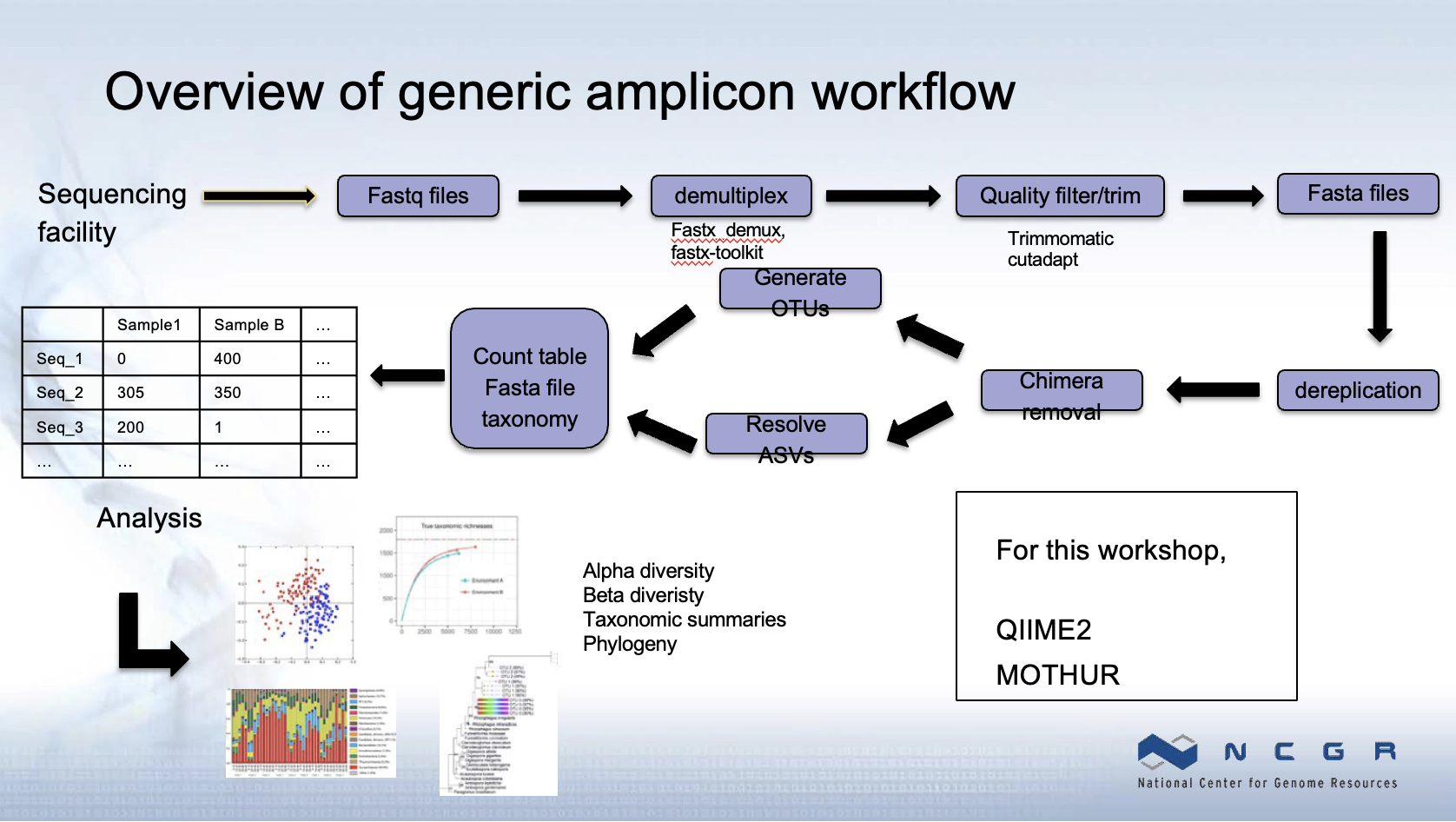
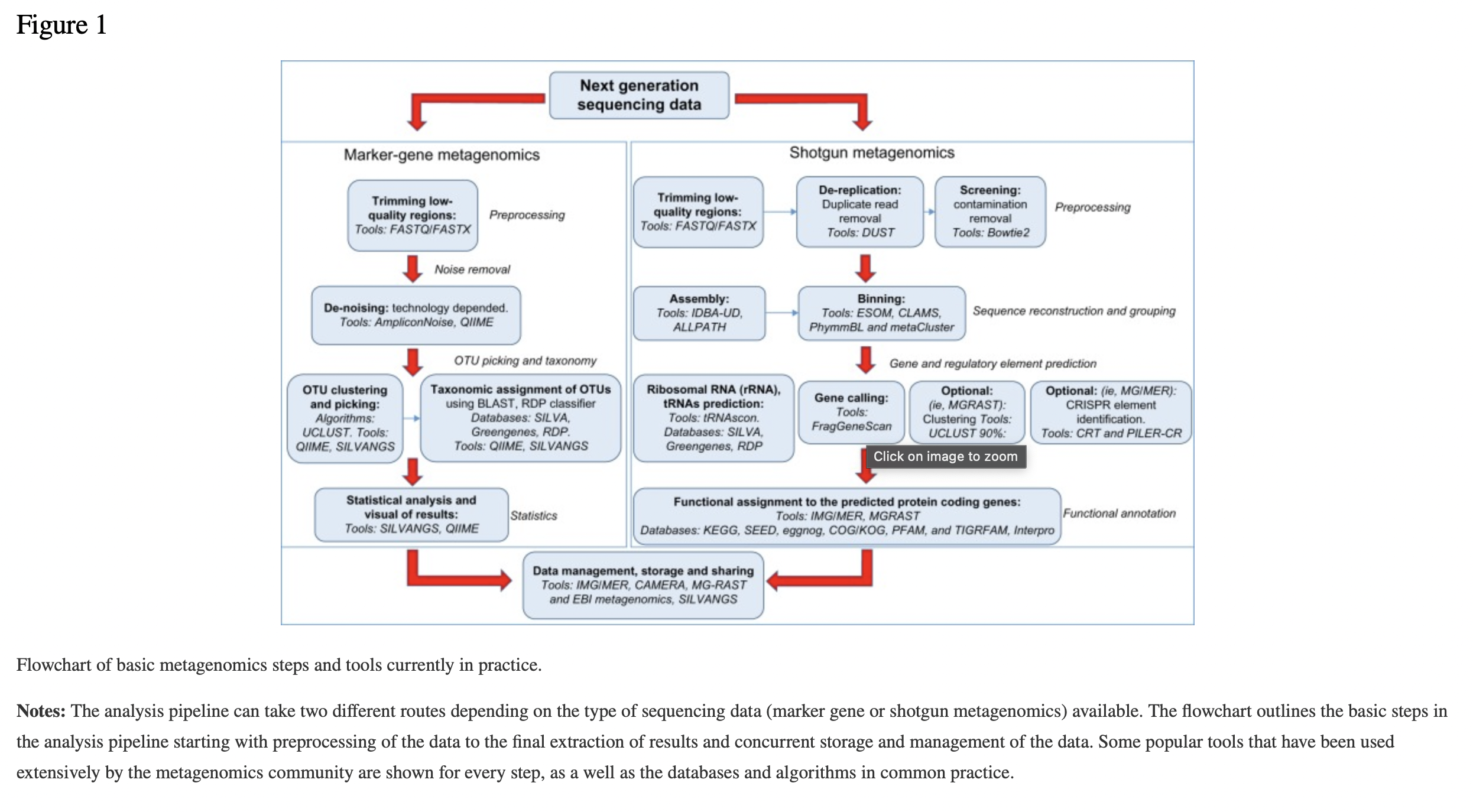
4.6.7 Generic Analysis Workflow
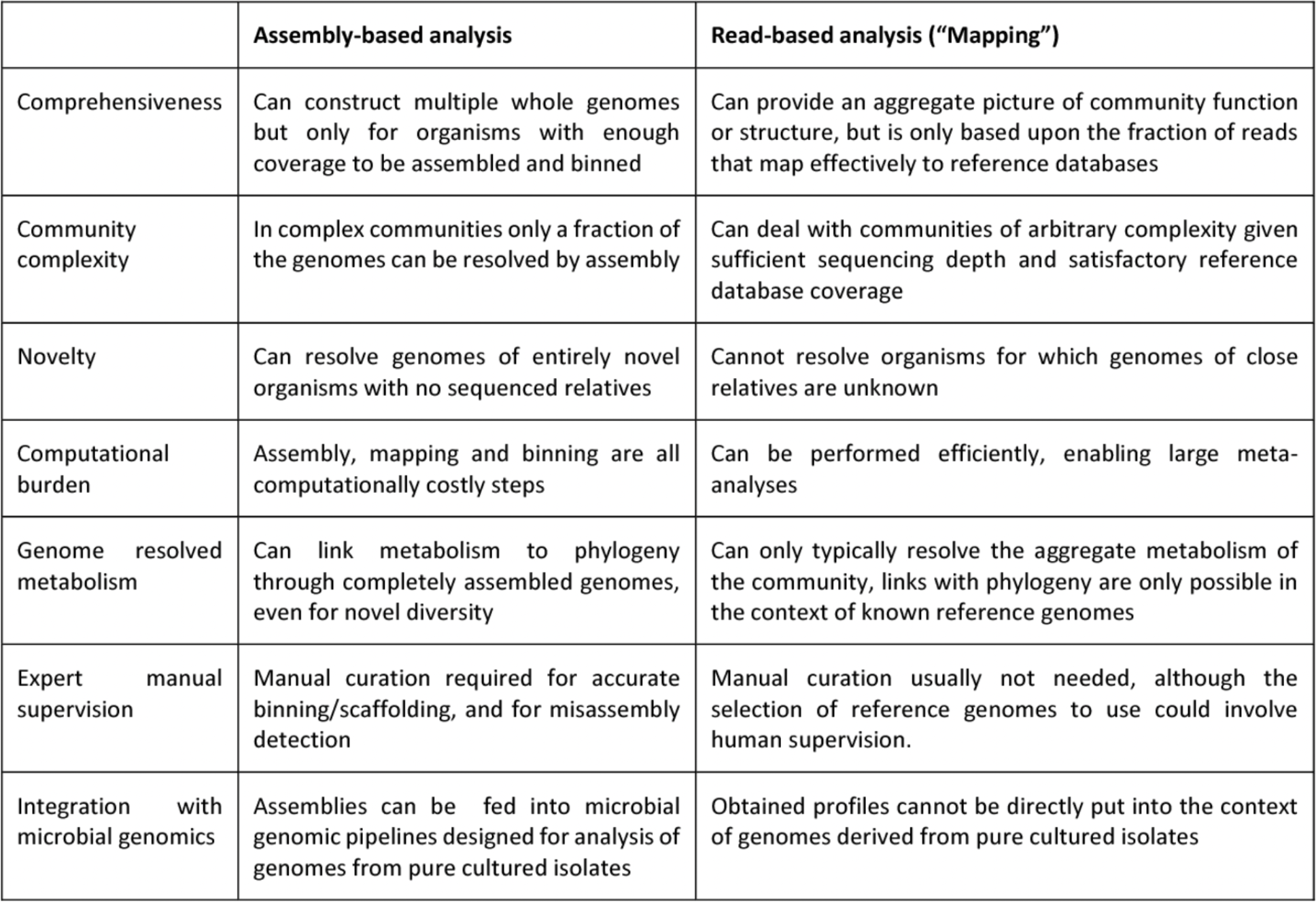
4.6.8 Strengths and weaknesses of assembly-based and read-based metagenomics analysis
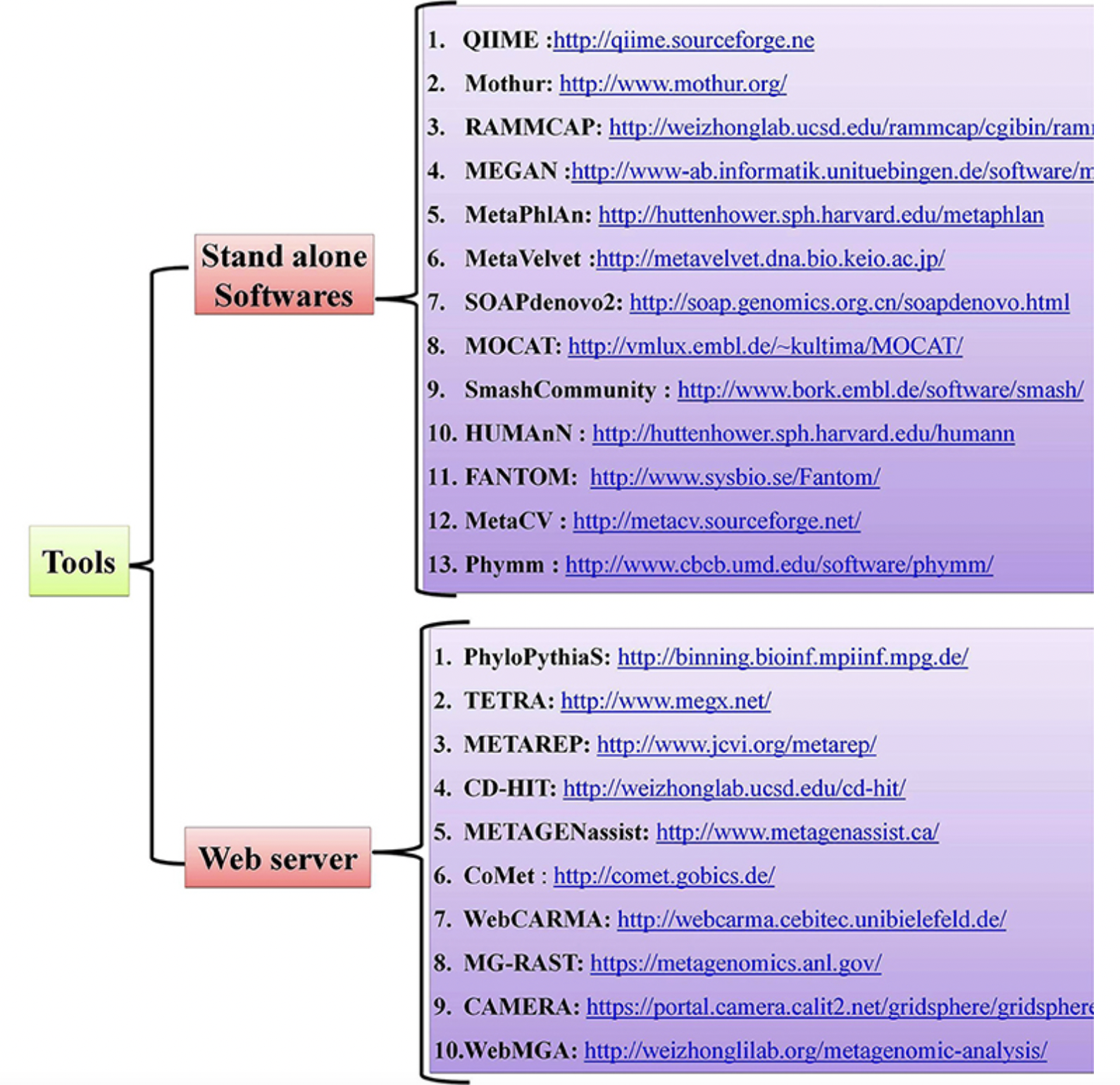
4.6.9 Tools and web servers related to gut microbiome studies.
4.6.10 Benefits and limitations of whole genome metagenomics
Benefits
- Integrative meta-omics
- Strain-level profiling
- Longitudinal study design
- Capability of sequencing large regions or entire genome
- Identification of organisms in addition to bacteria, archaea
- Increased prediction of genes and functional pathways
Limitations
- Expensive
- Compute intensive
- Incomplete databases
- Biases in functional profiling
- Unvalidated data in the public space
- Live or dead dilemma
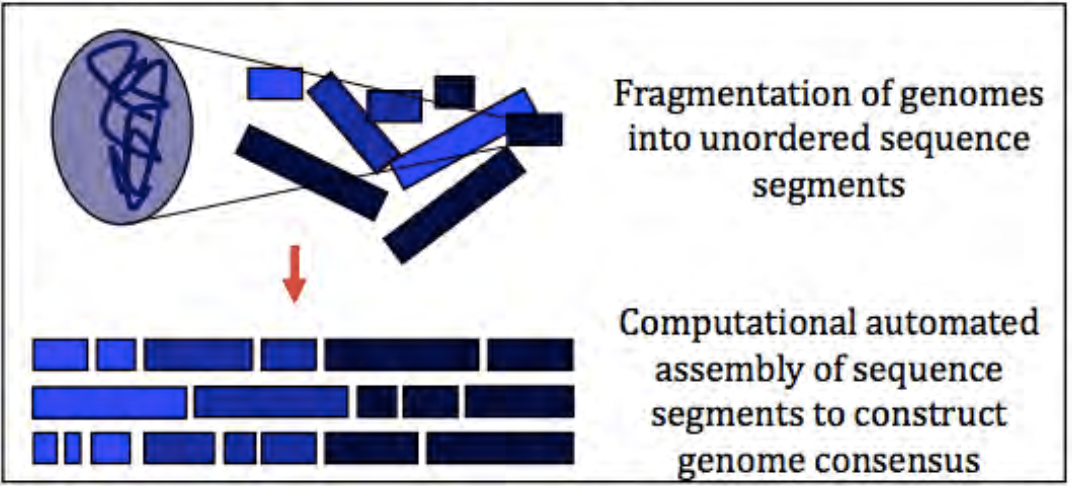
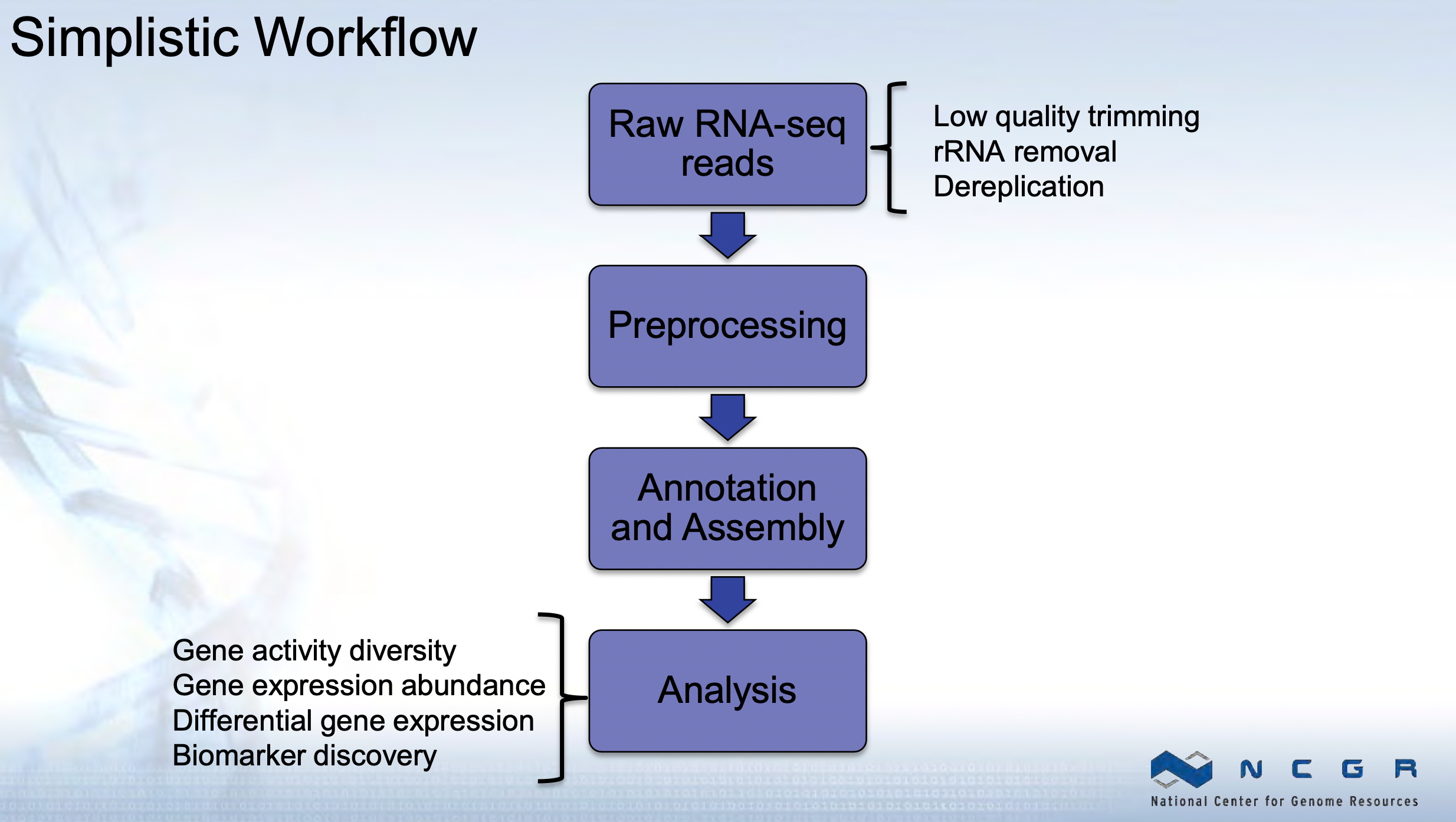
“What are they doing?” - Metatranscriptomics