Chapter 8 Multiple Sequence Alignment
8.1 SARS-Cov-2 variant genomes
We are going to line up some SARS-CoV-2 genomes so that their corresponding nucleotides all match up and their genes start and stop in the same place. This will involve adding in gaps here and there where some sequences have insertions relative to other sequences. We’ll start with a file that has genome sequences for some SARS-CoV-2 variants.
Do the following: On the server, create a directory called sars-variants in your home directory. Navigate into that directory Soft link to the file /home/data/nise/sars-cov-2-variants.fasta
Click for Answers
mkdir ~/sars-variants
cd ~/sars-variants
ln -s /home/data/nise/sars-cov-2-variants.fasta .
How many sequences are there in the file? Which SARS-CoV-2 variants are there?
Click for Answers
grep -c '>' sars-cov-2-variants.fasta
grep '>' sars-cov-2-variants.fasta
> Original variant (Wuhan-Hu-1)
> Alpha
> Delta
> Omicron
We’ll start with a java tool which will allow us to do an alignment and to visualize it.
Dowload the sars-cov-2-variants.fasta file onto your computer.
Install Jalview (https://www.jalview.org/). Click on download and choose the windows or mac version and install it.
Open up Jalview.
Close all the windows except the blank window at the back.
Go to main menu at the very top. Choose File > Input Alignment > From File - Then choose the sars-cov-2-variants.fasta file.
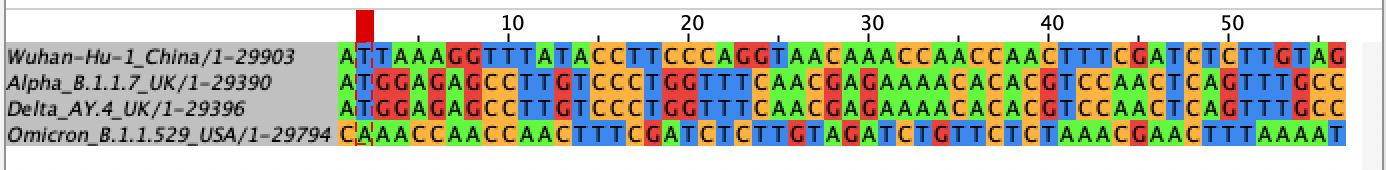
It is expecting an aligned fasta file and we are opening up a fasta file that hasn’t been aligned yet. Let’s take a look at it in its unaligned form, first.
Color the sequences by nucleotide by choosing: Colour > Nucleotide

Alpha and Delta line up pretty well but the other two are way off because their sequence is shifted relative to Alpha and Delta.
So, let’s align them now. We’ll use the MAFFT (Multiple Alignment using Fast Fourier Transform) aligner, which is a very fast multiple sequence aligner.
Choose: Web Service > Alignment > Mafft with defaults
Go ahead and color by nucleotide again.

Now the original (Wuhan-Hu-1) and the Omicron are lining up. The dashes mean that there is no sequence there. In other words, not all the variant genomes were sequenced and assembled all the way to the ends of the RNA genome molecule. Scroll to the right until you can see sequence in all four variants.

They all start to look identical here. The consensus and occupancy tracks can tell you quickly whether everything has sequence in that region (occupancy) and whether they all have the same nucleotide (consensus).
Scroll all the way to the right to see the end of the chromosome. Did every genome assembly make it to the end?
Click for Answers
No, they didn’t.

Take a look at the overview window. View > Overview
Expand it by hovering over the bottom right-hand corner until the cursor changes, then click and drag to make it bigger.
This has all four genomes one on top of each other across the whole genome sequence (~30kb).
If you click on the overview, it will take you to the corresponding position in the alignment window. Try to find a few regions on the overview that don’t look the same in all four variant genomes. Click on them and see what they look like in the alignment window.
Let’s look for indels (insertions and deletions), ignoring those on the ends of the chromosome that are likely due to lack of sequence coverage rather than being a true indel event.
Close the overview window. Scroll through the genome looking for indels. These will be marked by dashes and both the consensus and occupancy tracks will not be full height. Note the following for each indel:
start position
length
variant(s) that are different from Wuhan-Hu-1
insertion or deletion compared to Wuhan-Hu-1Compare with each other to make sure you got them all. What do you notice? What might be the significance of your observations?
Click for Answers
Start Length Variant(s) Type
11288 9 Alpha Deletion
21633 9 Omicron Deletion
21765 6 Alpha/Omicron Deletion
21991 3 Alpha Deletion
22029 6 Delta Deletion
28248 6 Delta Deletion
28271 1 Alpha/Delta Deletion
28362 9 Omicron DeletionPossible answers:
Lengths They are all multiples of 3 except for 1. This avoids frameshifts. In other words, if the indel is a multiple of 3, you gain or loss an amino acid or two or three but the downstream amino acids would stay the same.
Positions They seem to be clustered in a couple of different regions of the genome. It might mean that the indels that have selective advantage are only in a couple of genes.
Type They are all deletions. There are some insertions seen in other variants but they are much more rare compared to deletions.
Use the coordinates below to figure out what genes the indels are in.
“NSP1”,“266-805”, “NSP2”,“806-2719”, “NSP3”,“2720-8554”, “NSP4”,“8555-10054”, “NSP5”,“10055-10972”, “NSP6”,“10973-11842”, “NSP7”,“11843-12091”, “NSP8”,“12092-12685”, “NSP9”,“12686-13024”, “NSP10”,“13025-13441”, “NSP11”,“13442-13480”, “NSP12”,“13442-13468|13468-16236”, “NSP13”,“16237-18039”, “NSP14”,“18040-19620”, “NSP15”,“19621-20658”, “NSP16”,“20659-21552”, “Spike”,“21563-25384”, “NS3”,“25393-26220”, “E”,“26245-26472”, “M”,“26523-27191”, “NS6”,“27202-27387”, “NS7a”,“27394-27759”, “NS7b”,“27756-27887”, “NS8”,“27894-28259”, “N”,“28274-29533”, “NS9b”,“28284-28577”, “NS9c”,“28734-28955”
Click for Answers
Start Length Variant(s) Type Gene
11288 9 Alpha Deletion NSP6
21633 9 Omicron Deletion Spike
21765 6 Alpha/Omicron Deletion Spike
21991 3 Alpha Deletion Spike
22029 6 Delta Deletion Spike
28248 6 Delta Deletion NS8
28271 1 Alpha/Delta Deletion non-genic
28362 9 Omicron Deletion NS9b
Now let’s do a multiple sequence alignment on the server. This will allow us to do much bigger datasets.
Log into the server Enter a screen Go into your ~/sars-variants directory
Activate the environment.
conda activate msa-treemafft sars-cov-2-variants.fasta > mafft.sars-cov-2-variants.fasta
Take a look at the file and make sure it looks as expected.
8.2 Spike protein
You can also do alignments of protein sequences. Working on the protein level shows you missense and nonsense mutations, which are the main drives of phenotypic change. Any silent mutations (mutations that don’t change the amino acid sequence) are missed. While silent mutations only affect phenotype very rarely, they are useful to see how closely related organisms are evolving and, in the case of pathogens, they can inform modes of transmission. So, working on the protein vs the DNA level each have their advantages.
GISAID pulls out the spike protein sequences from all of the genomes. So we don’t all download our own copy of the spike proteins and use up space, I’ve downloaded a copy (updated 7/2/2023).
Create a soft link to /home/data/nise/spikeprot230702.fasta.gz We can continue to work in the sars-variants directory.
Click for Answer
ln -s /home/data/nise/spikeprot230702.fasta.gz .
How many sequences are there?
Click for Answer
zgrep -c '>' spikeprot230702.fasta.gz
Check out the headers to see their format
Click for Answer
zgrep '>' spikeprot230702.fasta.gz | head
Get the sequences for Denmark, Norway, and Sweden.
Note: Because each sequence has only one sequence line you just need to grep the appropriate headers plus the line that follows each header (grep -A 1). Unfortunately, when grep is skipping lines it adds in an extra line with “–”. You’ll need to also get rid of those lines after you grep (grep -v ‘--’).
Note: the dashes have to be escaped with backslashes so that grep reads them as literal dashes.
Then gzip the file to compress it and save space.
Click for Answer
zgrep -A 1 -E "Denmark|Norway|Sweden" spikeprot230702.fasta.gz | grep -v '\-\-' > dns.spikeprot230702.fasta
gzip dns.spikeprot230702.fasta
OR
zgrep -A 1 -E "Denmark|Norway|Sweden" spikeprot230702.fasta.gz | grep -v '\-\-' |gzip > dns.spikeprot230702.fasta.gz
We need to add in Wuhan-Hu-1 as our outgroup. We’ll use an early isolate from the US that matched the Wuhan-Hu-1 that is present in the spike sequence file. Get isolate EPI_ISL_759860’s header and sequence from spikeprot230702.fasta.gz and put it into a file called wuhanhu1.fasta.
Note: There are several accessions that start with EPI_ISL_759860 but we want EPI_ISL_759860 not EPI_ISL_7598607, for instance, so search for ‘EPI_ISL_759860|’
Click for Possible Answers
zgrep -A1 'EPI_ISL_759860|' spikeprot230702.fasta.gz > wuhanhu1.fasta
Now combine the dns.spikeprot230702.fasta (Denmark, Norway, Sweden spike sequences) and the wuhanhu1.fasta file you just made using the “cat” command. Put it in a file called dnsw.spikeprot230702.fasta
Click for Possible Answers
cat wuhanhu1.fasta dns.spikeprot230702.fasta > dnsw.spikeprot230702.fasta
Let’s make an EPI_SET for acknowledgement. See if you can grab all the EPI ids from the headers, put them in a file, and download them to your computer. Then go to GISAID and log in so you can make an EPI_SET. One way to make an EPI_SET is to click on search then EPI_SET (bottom right). Load your file of EPI ids.
Click for Answer
zgrep '>' dnsw.spikeprot230702.fasta | cut -f 4 -d '|' > dnsw.epi.txt
Go to GISAID to get your EPI_SET as described above.
The Wuhan-Hu-1 header is really long and has a lot of special characters. Since we will be feeding that in on the command line as the outgroup, let’s make it shorter.
Find the Wuhan-Hu-1 header.
Click for Possible Answers
grep '>' Wuhan dnsw.spikeprot230702.fasta
Let’s replace it so it just says Wuhan-Hu-1. Here is one way to do it.
NOTE: -i.bak tells sed to edit the file in place and make a backup of the original file, adding the .bak suffix.
sed -i.bak -e 's/^>.*\(Wuhan-Hu-1\).*/>\1/' dnsw.spikeprot230702.fastaMake a multiple sequence alignment. MAFFT should be able to automatically detect that it is a protein sequence.
Since this is is bigger dataset, let’s parallelize it using the –threads parameter. Give it 10 threads, which will split the job up into 10 pieces to be run simultaneously before bringing everything back together.
Click for Answer
mafft --thread 10 dnsw.spikeprot230702.fasta > mafft.dnsw.spikeprot230702.fasta
Download the multiple sequence alignment and look at it in Jalview.
Now repeat this for your three countries.
8.3 Arcturus Genomes
Finally, we’ll make an MSA from the Arcturus genomes. These are whole genome alignments of a lot more genomes so it will take a little longer. Make sure you are in a screen.
Go to this directory: ~/gisaid_genomes We’ll use the dns.arcturus.fasta file that you made earlier.
Click for Answers
mafft --thread 10 dns.arcturus.fasta > mafft.dns.arcturus.fasta
Download it and look at it in Jalview.
8.4 Omicron Recombinant Case study
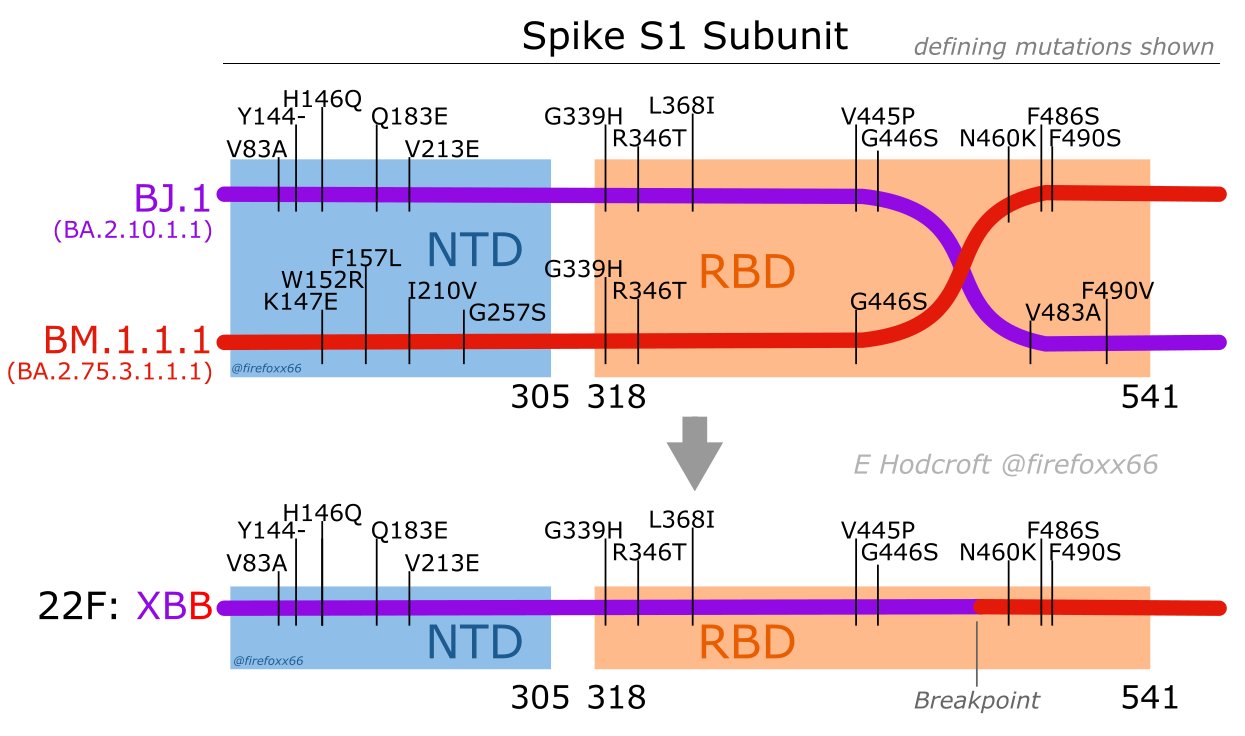
A new omicron subvariant arose from a recombination of two subvariants: BJ.1 (BA.2.10.1.1 in the 21L omicron lineage and BM.1.1.1 (BA.2.75.3.1.1.1) in the 22D omicron lineage (https://covariants.org/variants/22F.Omicron). It likely arose in mid-2022 from a crossover event within a person whose cells were simultaneously infected by BJ.1 and BM.1.1.1. The new variant was named XBB (the “X” stands for the crossover event that occured between the two “B” subvariants; hence, XBB).
Create a soft-link to /home/data/nise/recombinant.fa which has the two progenitor sequences and the XBB sequence. Or, rather, since the XBB spike protein sequence was hard to find, I grabbed the XBB.1 sequence. Note that the XBB.1 sequence has a G252V (G changed to V at position 252) mutation that XBB doesn’t have.
Create and view a multiple sequence alignments (MSA) using MAFFT and Jalview and see if you can narrow down where the crossover occured. You can do the MSA either on the command line or in Jalview.
After you have narrowed it down, click on the answer below and compare what you found to the picture. Note that the numbers in the picture below are based on the coordinates in the original SARS-CoV-2 virus. The three sequences that you are working with all have 3 amino acids that are deleted near the beginning of their sequence, thereby shifting coordinates by 3 (ie. 83 in the picture will show up as position 80 in your alignment).
Click for Answer